
Abstract
Gametogenetin-binding protein 2 (GGNBP2) is encoded in human chromosome 17q12-q23, a region known as a breast and ovarian cancer susceptibility locus. GGNBP2, also referred to ZFP403, has a single C2H2 zinc finger and a consensus LxxLL nuclear receptor-binding motif. Here, we demonstrate that GGNBP2 expression is reduced in primary human breast tumors and in breast cancer cell lines, including T47D, MCF-7, LCC9, LY2, and MDA-MB-231 compared with normal, immortalized estrogen receptor α (ERα) negative MCF-10A and MCF10F breast epithelial cells. Overexpression of GGNBP2 inhibits the proliferation of T47D and MCF-7 ERα positive breast cancer cells without affecting MCF-10A and MCF10F. Stable GGNBP2 overexpression in T47D cells inhibits 17β-estradiol (E2)-stimulated proliferation as well as migration, invasion, anchorage-independent growth in vitro, and xenograft tumor growth in mice. We further demonstrate that GGNBP2 protein physically interacts with ERα, inhibits E2-induced activation of estrogen response element-driven reporter activity, and attenuates ER target gene expression in T47D cells. In summary, our in vitro and in vivo findings suggest that GGNBP2 is a novel breast cancer tumor suppressor functioning as a nuclear receptor corepressor to inhibit ERα activity and tumorigenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Previous genetic studies identified that allelic loss in familial breast and ovarian cancer patients is observed on human chromosome 17 q12-q23, in which the well-known tumor suppressor breast cancer 1 (BRCA1) is located [1–4]. However, mutations in BRCA1 account for only ~10 % of all breast cancers [5, 6]. While numerous breast cancer susceptibility genes, i.e., BRCA1, BRCA2, PTEN, TP53, ATM, NBN, CHEK2, and PALB2 have been identified, the genes responsible for the remaining ~50 % of all breast tumors are not yet established [7].
In silico analyses of 693 genes encoded in human chromosome 17q12-q23 region revealed a gene ~6.3 MB upstream of BRCA1-encoding gametogenetin-binding protein 2 (GGNBP2), also known as zinc finger protein 403 (ZFP403) with additional alias ZNF403, DIF3, LZK1, LCRG1 [8]. GGNBP2 was originally identified as a dioxin-induced factor 3 (DIF3) in mouse embryonic stem cells in 2001 [9]. GGNBP2′s name comes from its interaction with a testicular protein gametogenetin [10]. GGNBP2 encodes a single C2H2 [Cystine–Cystine–Histidine–Histidine] zinc finger and has a consensus nuclear receptor (NR) binding box LxxLL [Leucine-x-x (any amino acid)-Leucine-Leucine] [11, 12]. LxxLL motifs are also found in other known breast cancer susceptibility genes, i.e., BRCA1, NCOA1 (also known as steroid receptor coactivator 1, SRC-1), and NCOA3 (also known as steroid receptor coactivator 3, SRC-3) [5, 13–15]. Therefore, we hypothesized that GGNBP2 may be a putative NR coregulator and a candidate breast cancer susceptibility gene.
GGNBP2 is evolutionarily conserved, sharing 87 % homology in nucleotide sequence and 96 % homology in amino acid sequence between humans and mice. Its mRNA transcripts have been detected in a number of tissues including testis, heart, brain, lung, liver, kidney, pancreas, placenta, and skeletal muscle [8, 9]. GGNBP2 encodes a full-length protein composed of 740 amino acids. The protein was reported to localize to the nucleus [9, 10]. In humans, a shorter form of GGNBP2 containing 288 amino acids at the N-terminus has been reported and named as laryngeal carcinoma-related gene 1 (LCRG1) because of its downregulation in primary laryngeal carcinomas. It has been suggested that LCRG1 may play a role in laryngeal tumorigenesis [8, 16, 17]. More recently, reduced GGNBP2 was observed in chemotherapeutic-resistant ovarian cancer cell lines [18]. However, the function of GGNBP2 in breast cancer and steroid hormone action remains unknown. Here, we provide evidence that GGNBP2 inhibits breast cancer cell growth through modulating estrogen receptor α (ERα) action.
Materials and methods
Multiple human tissue expression array assays
Multiple human tissue expression cDNA arrays from nine different adult normal tissues and pairwise tumor patient samples including the normal and breast cancer tissues of 50 individuals were purchased from BD Biosciences Clontech (San Jose, CA, USA) and hybridized with a random primed cDNA probe of human GGNBP2. The cDNA probe was labeled using the T7QuickPrime Kit (Amersham, Pittsburgh, PA, USA) and α-32P-dCTP according to the instructions of the manufacturer. The probe was purified using ProbeQuant G-50 columns (Amersham). After hybridization and washing, the hybridized membranes were scanned using a Molecular Dynamic Storm PhosphorImager (Molecular Dynamics, Sunnyvale, CA, USA) to capture the phosphoimages and detect the signal intensity at each spot. The membranes were then boiled in 0.1 % SDS to strip off hybridized GGNBP2 probe and re-hybridized with an α-32P-dCTP labeled human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA probe. Following background correction, GGNBP2 signal intensity of each sample on the membranes was normalized to the housekeeping gene reference GAPDH and then the signal intensity was calculated for each spot on the arrays.
Reverse transcription-polymerase chain reaction (RT-PCR) and real-time quantitative PCR (qPCR)
Total RNA was extracted from human breast cell lines and xenograft tumor tissues using TRIZOL Reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions. RNA samples from mammary glands of wild-type C57 mice at virgin and pregnancy day 4.5, 6.5, 12.5, and 18, and from normal mammary gland and mammary tumor of Wnt1 transgenic mice were generously provided by Dr. John Lydon (Baylor College of Medicine, Houston, TX, USA). Total RNA was adjusted to a concentration of approximately 1.0 µg/µL. Two microgram total RNA was reverse transcribed into cDNA with random primers (Invitrogen) and avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI, USA). The cDNA was amplified by PCR with the primer sets of human or mouse GGNBP2 and GAPDH, respectively. PCR primers, as listed below, were designed according to the sequences obtained from GenBank using the Vector NTI 12.0 program (Invitrogen) and synthesized by Operon Technologies (Alameda, CA, USA). Mouse Ggnbp2: 5′-CTCATTGGTGAACTTGACTGC-3′ (forward) and 5′-TCACTGCTTTCTCGTCTGCGGTG-3′ (reverse). Mouse Gapdh: 5′-TGCAGTGGCAAAGTGGAGAT-3′ (forward) and 5′-TTTGCCGTGAGTGGAGTCATA-3′ (reverse). For determination of the expression of exogenous GGNBP2 and amphiregulin (AREG) in stable cell lines, RT-PCR was performed using following primers (a forward primer 5′-CACCATGGGTTCTCATCATCATCATCATC-3′, which recognizes the exogenous His C tag and a reverse primer 5′-AAACATCCATCCAAC ATCCTC-3′, which recognizes the human GGNBP2 cDNA). Human AREG: 5′-GTCTTCAGGGAGTGAGATTTCC-3′ (forward) and 5′-GTGAGGATCACAGCAGACATAA-3′ (reverse). All primers were designed to amplify the products that covered more than one exon. Each PCR-cycle consisted of denaturation for 45 s at 94 °C, annealing for 1 min at 57 °C, and extension for 1 min at 72 °C. The amplified products were separated by electrophoresis in agarose gels and stained using ethidium bromide.
Real-time qPCR analysis was also performed to determine endogenous GGNBP2 expression in MCF-10A, MCF-7, T47D, LCC9, LY2, and MDA-MB-231 cells and CCND1 and TFF1 expression in T47D cell xenograft tumor tissues using the predesigned TaqMan probes and primers (Invitrogen).
Cell lines
Human breast cell lines T47D, MCF-7, MCF-10A, MCF-10F, and MDA-MB-231 cells, and HeLa cells were purchased from ATCC (Manassas, VA, USA). LCC9 and LY2 cell lines were derived from MCF-7 cells by cultivation with the antiestrogens ICI 182,780 (Fulvestrant) and LY 117018, respectively, and were graciously provided as a gift by Dr. Robert Clarke, Georgetown University [19]. All the cells were maintained in the recommended culture media: RPMI-1640 medium (Sigma, St Louis, MO, USA) for T47D cells; ATCC-formulated Eagle’s Minimum Essential Medium (DMEM, ATCC) for MCF7, LCC9, and LY2 cells; MEGM mammary epithelial cell growth medium from Lonza (Allendale, NJ, USA) for MCF10A cells, L-15 for MDA-MB-231 cells (Sigma); and a mixture of DMEM and F12 medium (Sigma) for MCF-10F and HeLa cells, respectively. All culture media were supplemented with 10 % fetal bovine serum (FBS, Sigma) and antibiotic–antimycotic solution (Invitrogen). All cells were cultured at 37 °C in a 5 % CO2 in air atmosphere.
DNA transfections and generation of cell lines with stable overexpression of exogenous GGNBP2
The human GGNBP2 cDNA fragment was obtained by PCR using human testis cDNA as the template and validated by DNA sequencing. The correct full-length human GGNBP2 cDNA was cloned into pcDNA3–6HisC vector (Invitrogen) to construct the mammalian expression plasmid pcDNA3–HisC tagged-GGNBP2 (pcDNA3–GGNBP2). To generate stable GGNBP2 overexpression cells, T47D, MCF-7, MCF-10A, and MCF-10F cells were cultured in their perspective culture medium to approximately 50 % confluence in a 12-well plate and were transfected with 1 µg of either pcDNA3–GGNBP2 or pcDNA3–HisC control plasmid using Lipofectamine 2000 (Invitrogen). The cells were then cultured in the same medium containing 200 µg/mL G418 (Cellgro molecular genetics, Manassas, VA, USA) for 21 days. Monoclonal resistant cells were obtained using limiting dilution assay, maintained in medium supplemented with G418 (200 mg/L) for amplification, and subcultured for subsequent experiments. The stable transfection and expression of GGNBP2 in the T47D, MCF-7, MCF-10A, and MCF-10F cells were verified by RT-PCR analysis.
Cell proliferation assays
The in vitro cell proliferation assays were performed by cell counting using a hemocytometer under the microscope, and by the measurement of the absorbance at 490 nm using the CellTiter 96 AQueous One Solution cell proliferation assay (MTS) kits following the protocol recommended by the manufacturer (Promega). For cell-counting studies, equal number of control T47D (T47D-HisC) and GGNBP2-overexpressing cells (T47D-GGNBP2 clones #12 and #15) were seeded on a 24-well plate (2.5 × 104 cells/well), and cultured for 2, 4, 6, 8, and 10 days. Media were changed with fresh media every other day. At the denoted days of culture, cells were harvested using trypsin solution, and cell numbers were determined using a hemocytometer under an Olympus light microscope. Eight samples per each group were counted at each time point. To determine the absorbance at 490 nm, stably transfected T47D, MCF-7, MCF-10A, and MCF-10F cells were seeded in 96-well plates at an initial density of 8 × 103 cells/well suspended in their perspective media and cultured for 4 days. Then, these cells were incubated with CellTiter 96 AQueous One Solution at 37 °C for 1 h and the absorbance at 490 nm was determined using a Bio-Rad microplate reader (Hercules, CA, USA).
For the study of estradiol effects on T47D cell proliferation, equal numbers (8 × 103 cells/well in the 96-well plate) of control T47D-HisC and GGNBP2-overexpressing cells (clone #15) were cultured in medium supplemented with charcoal-extracted FBS (Sigma) for 24 h and then treated with various concentrations of 17β-estradiol (E2, Sigma) or ethanol. The medium was changed every 48 h. Cell proliferation assays were performed on day 5 after E2 treatment using a CellTiter96 AQueous One Solution Cell Proliferation kit (Promega).
Three-dimensional (3-D) cultures of T47D cells
Three-D Matrigel cell cultures were conducted as described previously [20]. In brief, parental T47D cells and GGNBP2-overexpressing T47D cells clone #15 were resuspended in chilled 100 % Matrigel (BD Biosciences, San Jose, CA, USA), and seeded on 24 well culture plates (5000 cells/well). These cells in the Matrigel were cultured in RPMI-1640 medium supplemented with 10 % FBS, and media were carefully replaced with fresh media every other day. At day 2, 4, and 6 after culture, the morphology of cultured cells in the matrigel was captured using an inverted phase-contrast microscope, and the cells were harvested by trypsinization. Cell number was determined using a hematocytometer under an Olympus light microscope.
Colony formation on soft-agar plates
Colony formation on soft-agar plates was performed as described previously [21]. Briefly, equal numbers of T47D-GGNBP2 cell clones #12, #15, and T47D-HisC cells (2 × 104 cells/35 mm dish) were plated in 0.2 % agar onto 0.5 % solidified agarose in RPMI-1640 media supplemented with 10 % FBS and then cultured at 37 °C. Culture medium was changed every 3 days. After 17 days, the plates were fixed with methanol at −20 °C for 5 min and then stained with crystal violet. The resulting colonies were counted under an Olympus dissecting light microscope.
Wound-healing cell migration analysis
Wound-healing cell migration analyses were performed as described previously [22]. Briefly, T47D-GGNBP2 cells (Clone #12 and #15) and T47D-HisC control cells were seeded in 6-well plates at a concentration of 2 × 106 cells/mL in RPMI-1640 supplemented 10 % FBS. Cells were allowed to form a confluent monolayer before wounding. After 12 h culture, a strip of confluent cells was removed across the monolayer using a rubber policeman and then cultured in the same medium for 5 days. Images were taken under an Olympus inverted phase-contrast light microscope, and the gap distances were measured using Olympus MicroSuite software to determine the cell migration rates.
Matrigel chamber invasion assay
The ability of T47D-GGNBP2 cell clones #12 and #15, parental T47D, T47D-HisC cells for passing through matrigel was measured using BD BioCoat Matrigel Invasion chambers according to the manufacturer’s suggested protocol (BD Biosciences). In brief, equal numbers of parental T47D, T47D-HisC, and T47D-GGNBP2 cells (2.5 × 104 cell/chamber) suspended in 500 µL serum-free RPMI-1640 medium were loaded to the upper chambers. The upper chamber is a 6.5-mm diameter insert with 8 µm pores of Matrigel-coated polyethylene terephthalate membrane. The inserts were placed in 24-well plates containing the same medium supplemented with 10 % FBS as the chemoattractant. The chambers were incubated for 12 h at 37 °C. At the end of incubation, the cells in the upper surface of the membrane were carefully removed with cotton swabs, and cells invaded across the matrigel to the lower surface of the membrane were fixed with methanol and stained with hematoxylin and eosin. Five randomly selected fields (×10 objective) were photographed with an Olympus inverted light microscope, and the total numbers of invaded cells on the lower surface of the membrane were counted under an Olympus inverted light microscope.
Western blot analysis
Cells and xenograft tumor tissues were homogenized in an ice-cold lysis buffer containing protease inhibitors (Roche, Indianapolis, IN, USA). The protein concentrations were measured using the Bradford method (Bio-Rad). An equal quantity of proteins were separated by 10 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes. The membranes were then blocked with 3 % nonfat milk and incubated overnight with mouse anti-cyclin D1 (CCND1, 1:500), mouse anti-trefoil factor 1 (TFF1, 1:500; Invitrogen), mouse anti-ERα (1:200; Santa Cruz Biotech), and rabbit anti-GGNBP2 (1:3000; Pacific Immuno Corp, Ramona, CA, USA) antibodies, respectively. Peroxidase-conjugated anti-mouse IgG (1:2000, Vector Laboratories, Burlingame, CA, USA) was used as the secondary antibody. Immunoblotting signals were detected using the Amersham ECL plus Western blotting detection system (Amersham). All membranes were re-probed with anti-β-actin antibody (Sigma), which served as a loading control.
In vitro histidine pulled-down assay
TNT-coupled reticulocyte lysate system (Promega) was used for transcription/translation of His-tagged GGNBP2 and His-tagged LxxLL-deleted GGNBP2 (GGNBP2 LXLΔ) protein. Briefly, the wild-type pCNDA3-6HisC-GGNBP2 plasmid and QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) were used to delete LxxLL motif according to the protocol recommended by the manufacturer. The deletion of the sequence encoded for LxxLL motif was verified by DNA sequencing. Rabbit reticulocyte was mixed with 1 μg pCDNA3-6His–GGNBP2 or pCDNA3-6His–GGNBP2 LXLΔ plasmid DNA and T7 RNA polymerase, and incubated at 30 °C for 90 min in a water bath according to the manufacturer’s recommended protocol. 35S-labeled human ERα and ERβ proteins were also synthesized using Promega’s TNT-coupled rabbit reticulocyte lysate system in the presence of 35S-methionine (MP Biochemicals, Santa Ana, CA, USA), and pcDNA3-ERα and -ERβ expression plasmids which do not contain a histidine-tag sequence in the expression vector (generously provided by Drs. Ray Wu and Bert W. O’Malley, Baylor College of Medicine). 10 µL of in vitro-translated His-tagged GGNBP2 protein was incubated with 10 uL of 35S-labeled in vitro-translated human ERα or ERβ protein in the presence or absence of 10−6 M E2 at 4 °C with constant rotation for 8 h. Histidine pulled-down assays were performed using the Dynabeads His-tag isolation and pull-down kit (Invitrogen) according to the manufacturer’s instructions. For each reaction, 50 µL Dynabeads were added and incubated on a roller at room temperature for 15 min. The protein-bead complexes were pulled-down by magnetic precipitation, washed, and then separated on 10 % SDS-PAGE gels. A 1/3 input of S35-labeled ERα or ERβ was also loaded on SDS-PAGE gel as a positive reference. These SDS-PAGE gels were dried and exposed to X-ray films to visualize the binding of His-tagged GGNBP2 to ERα or ERβ.
Transfections and luciferase measurements
For transient transfections, T47D-HisC, T47D-GGNBP2 clone #15, and HeLa cells were plated in 24-well plates. When the cells reached 50 % confluence, T47D-HisC and T47D-GGNBP2 clone #15 cells were transfected for 10 h with 0.1 μg of a mixture of the estrogen response element (ERE)-firefly luciferase reporter and pCMV-Renilla luciferase constructs (Qiagen, Valencia, CA, USA) using Lipofectamine 2000 (Invitrogen) in serum-free RPMI-1640 medium. HeLa cells were transfected with ERE-luciferase reporter alone or cotransfected with both ERE-luciferase reporter and pcDNA3-ERα expression plasmids in serum-free DMEM medium. After transfection, the medium was replaced by fresh normal growth medium and culturing continued for another 24 h. On the day of hormonal treatments, the medium was changed to phenol red- and serum-free RPMI-1640 for T47D cells or DMEM for HeLa cells and the cells were treated with or without 10−9 M E2 for 24 h. The luciferase reporter activities were measured using a dual-luciferase reporter assay kit and a luminometer from Promega according to the protocol provided by the manufacturer. To correct transfection efficiencies, the firefly luciferase activity was normalized to Renilla luciferase activity in the same cell lysates.
T47D cell xenograft
T47D-GGNBP2 and parental T47D cells (1 × 105/injection) were suspended in 100 μL PBS containing 50 % matrigel (BD Biosciences) and injected into the mammary fat pad of 4–5 week-old female nude mice (Vital River Company, Beijing, China). Tumor sizes were measured every 5 days in two dimensions using a caliper, and the tumor volume was calculated with the following formula: tumor volume (mm3) = 0.5 × ab 2 (a & b being the longest and shortest diameters of the tumor, respectively) as described previously [23]. Mice were sacrificed and the tumors were collected by the 28th day after T47D cell inoculation. Real-time qPCR was performed to validate the expression of GGNBP2 in the tumor xenografts. All mice were maintained as required under the National Institutes of Health guidelines for the care and use of laboratory animals. The use of animals in this study has been approved by the animal care and use committee of Tianjin cancer hospital.
Statistical analysis
Comparisons of means among more than two groups were performed by one-way analysis of variance (ANOVA). Student t test was used when comparing means of two groups. A p value <0.05 was considered as statistically significant.
Results
Ubiquitous expression of GGNBP2 in human tissues with the highest expression in mammary glands
A multiple tissue expression array was used to examine GGNBP2 expression in human tissues. Quantitative analyses by normalizing GGNBP2 relative to the housekeeping gene GAPDH showed that GGNBP2 expression was detected in all nine tissues examined with the highest levels in normal breast tissues and the lowest levels in normal kidney tissues (Fig. 1a). We also performed semi-quantitative RT-PCR analyses on multiple human cDNAs (purchased from Clontech) and found similar results (data not shown). RT-PCR also demonstrated that Ggnbp2 is expressed in mouse mammary glands with the highest levels in late pregnant and lactating mammary glands (Fig. 1b).

GGNBP2 expression is high in human breast, increased in mouse mammary gland in late pregnancy and lactation, and lower in breast tumors and cell lines than normal breast tissue and cell lines. a GGNBP2 expression in normal human tissues by dot blot analyses on Clontech multiple human tissue expression arrays reveals the highest expression of GGNBP2 in normal human breast tissue. Values are the mean ± SEM of 6 samples. b Ggnbp2 expression in normal mammary glands of virgin (V, 5 week-old), pregnant, and lactating mice and mammary tumors from Wnt1 transgenic (TG) mice by RT-PCR. Gapdh serves as an internal control and H2O is used as a negative control for RT-PCR. The Ggnbp2 blot is a longer exposure than the Gapdh blot to show the very low levels of Ggnbp2 mRNA detected in day 6.5 pregnant and Wnt1TG (Wnt1 transgenic) mouse mammary tumors. c Representative GGNBP2 expression in 15 breast cancer patients (indicated by numbers). N normal; T tumor; M metastatic cancer samples of the same patient (boxed). d Quantitative results of GGNBP2 mRNA levels in 50 paired normal and breast cancer tissues show a significant reduction of GGNBP2 expression in human breast cancer samples. N, normal; T, tumor. In a and d, data are the normalized housekeeping gene GAPDH levels. Values are the Mean ± SEM of 50 samples. b P < 0.01 compared to normal breast tissue samples. e qPCR using TaqMan primer/probe sets for determining relative endogenous GGNBP2 mRNA levels was normalized to 18S. Values are the average of triplicate determinations ± SEM of one experiment. CT values for GGNBP2 in LY2 and MDA-MB-231 breast cancer cell lines were ‘underdetermined’. a p < 0.05 compared to MCF-10A
Reduced GGNBP2 expression in human and mouse breast cancer samples and human breast cancer cell lines
A cancer expression array containing 50 pairwise human normal and breast tumor tissues was hybridized with radiolabeled probes to examine GGNBP2 expression in breast cancer samples (Fig. 1c). The reduction was also observed in metastatic cancer samples of the same patients (Fig. 1c). The hybridization intensity of GGNBP2 was noticeably lower in 45 breast tumor samples among these 50 patients examined, suggesting that aberrant expression of GGNBP2 in human breast cancer is fairly common. Quantitative analyses by normalizing to GAPDH showed that GGNBP2 expression levels were significantly reduced (Fig. 1d). Compared to normal, immortalized human breast cell line MCF-10A, GGNBP2 expression in six human breast cancer cell lines decreased at different degrees but significantly which included ERα positive cells lines MCF-7 and T47D, MCF-7-derived estrogen resistant cell lines LCC9 and LY2, and triple negative cell line MDA-MB-231 (Fig. 1e). The Wnt1 transgenic mice are known to spontaneously develop mammary gland tumors [24]. RT-PCR revealed that Ggnbp2 mRNA levels were dramatically reduced in the mammary tumors from Wnt1 transgenic mice (Fig. 1b).
Overexpression of GGNBP2 inhibited cell proliferation in ERα positive human breast cell lines
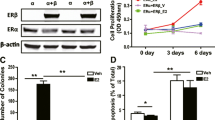
To address the potential antibreast tumor activity of GGNBP2 in vitro, we expressed exogenous His-tagged human GGNBP2 in T47D, MCF-7, MCF-10A, and MCF-10F cells (Fig. 2a). Semi-quantitative RT-PCR analysis confirmed the expression of exogenous GGNBP2 in these stably transfected cells (Fig. 2a). Western blot analysis showed that GGNBP2 protein levels were elevated in these stably transfected cells (Fig. 2a). Having generated these stable cells, we first examined the effect of overexpression of exogenous GGNBP2 on cell proliferation. As shown in Fig. 2b, the number of cells of GGNBP2-overexpressing ERα positive T47D and MCF-7 cells but not ERα negative MCF-10A and MCF-10F cells [25–27] was significantly reduced after 4 days of culture, when compared to corresponding empty vector transfected (HisC) cells (Fig. 2b). The results also show that stable expression of exogenous GGNBP2 significantly repressed T47D cell proliferation (Fig. 2c). To further determine whether GGNBP2 inhibited breast cancer cell proliferation by depending on ER, the proliferation of T47D cell stably overexpressing-GGNBP2 and T47D-HisC control cells were compared with or without E2 from 10−12 to 10−6 M (Fig. 2d). GGNBP2 overexpression rendered the cells unresponsive to E2-induced cell proliferation (Fig. 2d).

Stable expression of exogenous GGNBP2 inhibits the growth of human breast cell lines. a Representative RT-PCR (a) and Western blot (b) pictures of colonies formed in untransfected control (C) and stable (S) GGNBP2-transfected T47D, MCF-7, MCF-10A, and MCF-10F cells. The housekeeping gene β-actin (ACTB) serves as an internal control for RT-PCR and Western blotting. b Overexpression of GGNBP2 significantly inhibits cell proliferation, measured by MTS assays, of T47D and MCF-7 cell lines but not MCF-10A and MCF-10F cell lines. a P < 0.05 compared to corresponding parental cells. c Cell growth is inhibited by stable overexpression of GGNBP2 in T47D human breast cancer cells clones #12 and #15. The inset shows semi-quantitative RT-PCR for GGNBP2 in parental or GGNBP2 stable cells with ACTB serving as an internal control for RT-PCR. a P < 0.05 and b P < 0.01 compared to T47D-HisC cells. d E2-stimulated T47D cell proliferation is inhibited in T47D-GGNBP2 clone #15 cells relative to parental T47D-HisC stable cells. a P < 0.05 compared to T47D-HisC cells without E2 treatment; b,c P < 0.01 compared to T47D-HisC cells with E2 treatments. e Representative images of T47D-GGNBP2 and parental T47D cells grown in 3-D cultures. f. Cell growth of T47D-GGNBP2 clone #15 is significantly diminished. a P < 0.05 and b P < 0.01 compared to parental T47D cells. For b, c, d, and f, values are the mean ± SEM of 3 separate experiments
To examine if GGNBP2 can inhibit T47D cell growth in the context of extracellular matrix, T47D cells were cultured on Matrigel. Results showed that T47D-GGNBP2 cells formed cell masses, similar to their parental T47D cells (Fig. 2e). However, the sizes in T47D-GGNBP2 cell masses were smaller than those of parental T47D cells at days 4 and 6 of culture. These cell masses were then dispersed by trypsin digestion and the cell number was counted using a hematocytometer. As shown in Fig. 2f, the cell number of T47D-GGNBP2 masses was significantly reduced after 4 days of culture.
Reduced migration and colony formation in T47D cells with stable GGNBP2 expression
To test if GGNBP2 has a role in regulating cell migration, we performed wound-healing assays. As shown in Fig. 3a, parental T47D and T47D-HisC cells almost completely migrated into the empty spaces after 5 days. In contrast, only a few cells migrated to the empty spaces in T47D-GGNBP2 clones #12 and #15 cells after 5 days (Fig. 3a). Quantitative analysis indicated that migration percentage was significantly reduced in T47D-GGNBP2 clones #12 and #15 cells after 5 days of culture (Fig. 3b). These results could be due to attenuated cell growth, reduced cell migration, or both by overexpression of GGNBP2 in T47D cells. To further confirm that overexpression of GGNBP2 can inhibit cell migration, we performed cell migration assays using Matrigel chemotactic chambers. As shown in Fig. 3c, d, there was a significant decrease in invaded cells in T47D-GGNBP2 clones #12 and #15 when compared to either parental T47D or T47D-HisC control cells, suggesting that GGNBP2 inhibited cell migration. To examine if GGNBP2 can inhibit anchorage-independent T47D cell growth, we performed soft-agar colony formation experiments. As shown in Fig. 3e, f, the total number of colonies in T47D-GGNBP2 cells was significantly less than those in both parental T47D and T47D-HisC cells. Together, these results suggest that GGNBP2 inhibited the tumorigenic potential of T47D cells in vitro.

GGNBP2 overexpression inhibits T47D cell migration, anchorage-independent growth, and invasion. a and b Representative photos (a) taken at day 0 and after 5 days show that GGNBP2 overexpression in clones #12 and #15 detains T47D cell migration in a wound-healing assay. Quantitative data (b) show that GGNBP2 overexpression significantly inhibits T47D cell migration. Scale bar 250 µm. c and d. Representative photos (c) and quantitative data (d) show that GGNBP2 overexpression represses T47D cell invasion through the pores of membranes in chemotaxis chambers. Scale bar 100 µm, a P < 0.05 compared to parental T47D and T47D-HisC cells. e and f. Representative photos of colonies formed (e) in parental T47D, T47D-HisC and T47D-GGNBP2 cells on soft-agar plates after 17 days of culture. Quantitative data (f) indicate a significant reduction of cell invasion in T47D cells overexpressing GGNBP2. Scale bar 200 µm, a P < 0.05 compared to either parental T47D and T47D-HisC cells. For b, d, and e, values are the mean ± SEM of 3 separate experiments
GGNBP2 interacted with ERα in vitro
Thus far our studies showed that GGNBP2 overexpression inhibited cell proliferation in ERα positive T47D and MCF-7 human breast cancer cell lines and E2-stimulated T47D cell proliferation. Given the putative conserved NR-binding motif in human GGNBP2, we hypothesized that GGNBP2 interacts with ER to modulate its activity. Thus we performed an in vitro protein–protein interaction assay using in vitro transcribed/translated His-GGNBP2 or His-GGNBP2LXLΔ and ERα. As shown in Fig. 4a, ERα was pulled-down by His-tagged GGNBP2 but not His-tagged GGNBP2LXLΔ. However, His-tagged GGNBP2 did not pull-down ERβ (Fig. 4a). Interestingly, more ERα was pulled-down by His-tagged GGNBP2 in the presence of 1 µM E2 than in the absence of E2 (Fig. 4a). These results suggest that the LxxLL domain of GGNBP2 is critical for interaction with ERα and that this interaction is increased by E2 in vitro.

GGNBP2 protein interacts with ERα and inhibits E2-ERα transcriptional activity and target gene expression. a In vitro transcribed/translated His-GGNBP2 wild type (WT) or with the LxxLL deletion (MT) and 35S-met-labeled ERα were incubated at 4 °C for 8 h with vehicle ethanol (EtOH) or 10−6 M E2 and interactions assayed by histidine pull-down assay. Histidine pull-down assays show that 35S-met-labeled ERβ does not interact with WT His-GGNBP2 either presence or absence of E2. b Stable GGNBP2 overexpression significantly attenuates E2-stimulated ERE-luciferase reporter activity in T47D cells (Clone #15). a P < 0.01 compared to T47D-HisC cells treated with EtOH. b P < 0.05 compared to T47D-HisC cells treated with 1 nM E2. c GGNBP2 inhibits E2-ERα activity in transiently transfected HeLa cells. HeLa cells were transfected with an ERE-luciferase reporter, Renilla luciferase, and the indicated expression plasmids and treated with 10 nM E2 for 24 h prior to dual luciferase assay. Data are mean ±SEM of 4 replicates in one experiment. d Western blot demonstrates that CCND1 (cyclin D1) and TFF1 (pS2) protein levels are reduced in both stable T47D-GGNBP2 expression clones #12 and #15 compared to either parental T47D or T47D-HisC stable cells. Values are CCND1/ACTB relative to expression in T47D parental cells. e RT-PCR (a) and qPCR (b) of AREG mRNA expression shows decreased AREG in T47D-GGNBP2 clones #12 and 15. b P < 0.01 compared to parental T47D and T47D-HisC cells. For b, c, and e(b), values are the mean ± SEM of 3 separate experiments
GGNBP2 modulating ERα action in T47D and HeLa cells
To investigate whether GGNBP2 can modulate ERα transcriptional activity, we transiently transfected T47D cells with pcDNA3-HisC control vector or pcDNA3-GGNBP2 expression vector with an ERE-reporter plasmid and performed luciferase reporter assays. As expected, E2-stimulated activation of the ERE-reporter was observed in T47D cells transfected with HisC control vector plasmid. However, transfection of pcDNA3-GGNBP2 inhibited E2-induced ERE activation by >50 % (Fig. 4b). The effect of GGNBP2 on ERα transcriptional activity was further verified in HeLa cells transfected with ERα. Transfection with the GGNBP2 expression vector inhibited E2-induced ERE-reporter activity in the ERα-transfected HeLa cells (Fig. 4c).
Cyclin D1 (CCND1), Trefoil factor 1 (TFF1, also known as breast cancer estrogen inducible protein and pS2), and amphiregulin (AREG) are three well-known estrogen responsive genes in T47D cells, and CCND1 is critical for breast cell proliferation [28–30]. To address whether GGNBP2 downregulates endogenous E2-regulated proteins/genes, we performed Western blot and RT-PCR analyses to examine their expression in stable control T47D-HisC cells and T47D-GGNBP2 clone #15. As shown in Fig. 4d, e, the protein levels of both cyclin D1 and TFF1 and AREG mRNA levels were reduced in GGNBP2-overexpressing T47D cells. Taken together, these results suggest that GGNBP2 is an ERα modulator that can physically interact with ERα and repress its transcriptional activity.
Effect of GGNBP2 on xenograft tumor growth of T47D cells in nude mice
The antiproliferative activity of GGNBP2 in T47D cells in vitro led us to investigate whether its antitumor efficacy would be maintained in vivo. T47D-GGNBP2 and parental T47D cells were injected into the mammary fat pad of intact adult female nude mice. As expected, tumors developed in both T47D-GGNBP2 and parental T47D cells xenograft mice; however, GGNBP2 overexpression substantially reduced tumor progression relative to parental T47D cell controls (Fig. 5a, b). The tumor sizes of T47D-GGNBP2 xenografts were significantly smaller than the parental T47D cell xenografts (Fig. 5a, b). RT-PCR was performed to further validate that GGNBP2 was indeed elevated in T47D-GGNBP2 cells and their tumor xenografts. As shown in Fig. 5c, there was 4.3-fold increase of GGNBP2 mRNA levels in GGNBP2-overexpressing cells than their parental T47D cells. Similarly, GGNBP2 mRNA levels were markedly elevated in xenograft T47D-GGNBP2 tumor tissues (~5-fold increase) compared to the parental T47D tumor tissues (Fig. 5d). Thus, GGNBP2 stable expression was maintained in the T47D tumor xenograft. We examined ERα protein levels in xenograft tumor tissues of parental T47D and T47D-GGNBP2 cells by Western blot in 5 tumors (Fig. 5e). ERα protein levels are significantly lower in T47D-GGNBP2 xenograft tumors than parental T47D cell xenograft tumors (Fig. 5f). qPCR also demonstrated that the expression of ERα target genes TFF1 (Fig. 5g) and CCND1 (Fig. 5h) were significantly reduced in xenograft T47D-GGNBP2 tumor tissues. Together, these in vivo studies suggest that GGNBP2 has an antibreast tumor activity.

In vivo tumor suppression effect of GGNBP2 expression in T47D cells on xenografted nude mice. a Gross appearance of the tumors of xenografted parental T47D and T47D-GGNBP2 cells after 28 days. Scale bar 1 cm. b Tumor growth curves of xenografted parental T47D and T47D-GGNBP2 cells in nude mice. c and d Quantitative RT-PCR demonstrates that GGNBP2 expression in T47D-GGNBP2 cells before (c) and after (d) xenografting are significantly higher than parental T47D cells. e and f ERα protein levels in xenograft T47D and T47D-GGNBP2 tumor tissues were measured by Western blot in 5 tumors (1–5) (e) and the results show that ERα protein levels are lower in the T47D-GGNBP2 xenograft tumors than parental T47D xenograft tumors (f). g and h The expression of ERα target genes, TFF1 (G) and CCND1 (H) are significantly downregulated in xenograft tumor tissues of T47D-GGNBP2 cells. a P < 0.05 and b P < 0.01 compared to parental T47D cells. For b–d and f–h, values are mean ± SEM of 5 mice in each group
Discussion
Loss of heterozygosity in human chromosome 17q12-q23 is frequently observed in breast and ovarian cancers [1–4]. Although the tumor suppressor gene BRCA1 has been mapped to this region, BRCA1 mutations account for a small portion of all breast cancers [5–7]. In this study, we demonstrated that GGNBP2, a gene located in the chromosome 17q12 region [8], is highly expressed in normal human breast and its expression is significantly reduced in the majority of human breast cancer samples and in the five human breast cancer cell lines examined. Further, overexpression of exogenous GGNBP2 in ERα positive T47D human breast cancer cells results in a significant inhibition of their tumorigenic potential in vitro and in vivo, including a decrease in cell proliferation, anchorage-independent growth, migration and invasion, and tumor growth in nude mice. Together, these observations suggest that GGNBP2 is a candidate of breast tumor suppressor. Consistent with this suggestion that GGNBP2 is a tumor suppressor, a short form of human GGNBP2 known as LCRG1 was previously reported to be reduced in laryngeal carcinomas [8, 16, 31]. More recently, GGNBP2 expression was found to be downregulated in cisplatin- and carboplatin-resistant ovarian cancer cells [18]. Forced expression of LCRG1 in HeLa cells suppressed proliferation, whereas knockdown of LCRG1 by siRNA promoted multiplication [16, 32]. However, a study reported that down-regulation of the long form of human GGNBP2 by shRNA but not the short form in Hela cells resulted in a repression of cell proliferation [17], suggesting that the different isoforms of human GGNBP2 might play different roles in regulation of cell proliferation. In this respect, future studies will be required to examine whether there is an isoform-dependent effect of GGNBP2 on breast cancer cell growth. Moreover, it will be important to determine whether genetic, including germline and somatic deleterious mutations, or epigenetic changes, including post-translational modifications on histone proteins, contribute to the repression of GGNBP2 expression in breast tumors.
Estrogens play a critical role not only in normal mammary gland development but also in the initiation and progression of breast cancers [33, 34]. Binding of estrogens to ERα induces conformational changes and nuclear translocation, ERE binding, and the recruitment of coregulators to regulate transcription of ERα target genes [35–38]. This hormone-driven genomic action of ERα is crucial in promoting proliferation of breast cancers [34, 39]. In this work, we demonstrate that GGNBP2 overexpression markedly represses cell proliferation of ERα positive breast cancer cells but not ERα negative normal, immortalized breast epithelial cells. More importantly, GGNBP2 directly interacts with ERα, substantially impedes E2-induced ERE activity and inhibited estrogen target gene expression in breast cancer cells. Our results also show that GGBBP2 overexpression in T47D cells reduced ERα protein levels. Taken as a whole, our findings suggest that GGNBP2 functions as an ERα corepressor to influence the expression of its target genes, such as CCND1, TFF1, and AREG, and ultimately to control breast epithelial cell proliferation. In the light of our findings, more in-depth studies are warranted to further understand the molecular mechanisms by which GGNBP2 interplays with ERα to regulate ERα activity in breast cancers.
The LxxLL domain was initially identified in proteins that bind the activation function-2 region of nuclear receptor ligand-binding domains [40]. Subsequently, this conserved sequence is shown to interact with many NRs [41]. Deletion of the LxxLL domain abolished GGNBP2 binding to ERα, implying that the LxxLL domain has a key role in NR regulation. In addition to the regulation of the NR signaling, there are numerous examples protein–protein interactions involving LxxLL motifs [41]. This raises the possibility that GGNBP2 may also interact with other NRs or transcription factors in addition to ERα to exert its cancer suppression action, which is required further investigation.
More than 75 % of all cases of breast cancers are ERα positive [42]. The identification and characterization of coregulators that modulate ERα activity are important steps in the understanding, not only of the underlying mechanisms that control the expression of estrogen target genes but also those that influence the initiation, progression, treatment, and clinical prognosis of breast cancers. In estrogen-dependent tumors, GGNBP2 underexpression would be expected to result in an increase in ERα function and thus possibly render these cells more sensitive to low levels of estrogens. Thus, reduction or loss of GGNBP2 would contribute to the increased growth and invasiveness of ERα positive cancer cells. Our findings suggest that GGNBP2 acts as an ERα corepressor and its downregulation in breast tumors could have clinical implications for prognosis and therapeutic options for a subset of breast cancer cases. Future studies will explore this speculation.
In summary, GGNBP2 was identified as a novel tumor suppressor which is downregulated in human breast tumors and cell lines. Our in vitro and in vivo results suggest that GGNBP2 may function as a nuclear receptor corepressor to inhibit ERα’s transcriptional activity and consequently inhibit tumorigenic potential of breast cancer cells. Future studies will be required to determine whether genetic and/or epigenetic alternations of this gene associate with human breast cancers and test for clinical usefulness of this gene in conjunction with other known genetic markers for early detection of breast cancer risk and enhancement of current treatment strategies used in ERα-positive breast cancers.
References
Narod SA, Feunteun J, Lynch HT, Watson P, Conway T, Lynch J, Lenoir GM (1991) Familial breast-ovarian cancer locus on chromosome 17q12-q23. Lancet 338(8759):82–83
Sato T, Akiyama F, Sakamoto G, Kasumi F, Nakamura Y (1991) Accumulation of genetic alterations and progression of primary breast cancer. Cancer Res 51(21):5794–5799
Phelan CM, Borg A, Cuny M, Crichton DN, Baldersson T, Andersen TI, Caligo MA, Lidereau R, Lindblom A, Seitz S, Kelsell D, Hamann U, Rio P, Thorlacius S, Papp J, Olah E, Ponder B, Bignon YJ, Scherneck S, Barkardottir R, Borresen-Dale AL, Eyfjord J, Theillet C, Thompson AM, Larsson C et al (1998) Consortium study on 1280 breast carcinomas: allelic loss on chromosome 17 targets subregions associated with family history and clinical parameters. Cancer Res 58(5):1004–1012
Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, Soucy P, Fredericksen Z, Barrowdale D, Dennis J, Gaudet MM, Dicks E, Kosel M, Healey S, Sinilnikova OM, Lee A, Bacot F, Vincent D, Hogervorst FB, Peock S, Stoppa-Lyonnet D, Jakubowska A, kConFab I, Radice P, Schmutzler RK, Swe B, Domchek SM, Piedmonte M, Singer CF, Friedman E, Thomassen M, Ontario Cancer Genetics N, Hansen TV, Neuhausen SL, Szabo CI, Blanco I, Greene MH, Karlan BY, Garber J, Phelan CM, Weitzel JN, Montagna M, Olah E, Andrulis IL, Godwin AK, Yannoukakos D, Goldgar DE, Caldes T, Nevanlinna H, Osorio A, Terry MB, Daly MB, van Rensburg EJ, Hamann U, Ramus SJ, Toland AE, Caligo MA, Olopade OI, Tung N, Claes K, Beattie MS, Southey MC, Imyanitov EN, Tischkowitz M, Janavicius R, John EM, Kwong A, Diez O, Balmana J, Barkardottir RB, Arun BK, Rennert G, Teo SH, Ganz PA, Campbell I, van der Hout AH, van Deurzen CH, Seynaeve C, Gomez Garcia EB, van Leeuwen FE, Meijers-Heijboer HE, Gille JJ, Ausems MG, Blok MJ, Ligtenberg MJ, Rookus MA, Devilee P, Verhoef S, van Os TA, Wijnen JT, Embrace Hebon, Frost D, Ellis S, Fineberg E, Platte R, Evans DG, Izatt L, Eeles RA, Adlard J, Eccles DM, Cook J, Brewer C, Douglas F, Hodgson S, Morrison PJ, Side LE, Donaldson A, Houghton C, Rogers MT, Dorkins H, Eason J, Gregory H, McCann E, Murray A, Calender A, Hardouin A, Berthet P, Delnatte C, Nogues C, Lasset C, Houdayer C, Leroux D, Rouleau E, Prieur F, Damiola F, Sobol H, Coupier I, Venat-Bouvet L, Castera L, Gauthier-Villars M, Leone M, Pujol P, Mazoyer S, Bignon YJ, Collaborators GS, Zlowocka-Perlowska E, Gronwald J, Lubinski J, Durda K, Jaworska K, Huzarski T, Spurdle AB, Viel A, Peissel B, Bonanni B, Melloni G, Ottini L, Papi L, Varesco L, Tibiletti MG, Peterlongo P, Volorio S, Manoukian S, Pensotti V, Arnold N, Engel C, Deissler H, Gadzicki D, Gehrig A, Kast K, Rhiem K, Meindl A, Niederacher D, Ditsch N, Plendl H, Preisler-Adams S, Engert S, Sutter C, Varon-Mateeva R, Wappenschmidt B, Weber BH, Arver B, Stenmark-Askmalm M, Loman N, Rosenquist R, Einbeigi Z, Nathanson KL, Rebbeck TR, Blank SV, Cohn DE, Rodriguez GC, Small L, Friedlander M, Bae-Jump VL, Fink-Retter A, Rappaport C, Gschwantler-Kaulich D, Pfeiler G, Tea MK, Lindor NM, Kaufman B, Shimon Paluch S, Laitman Y, Skytte AB, Gerdes AM, Pedersen IS, Moeller ST, Kruse TA, Jensen UB, Vijai J, Sarrel K, Robson M, Kauff N, Mulligan AM, Glendon G, Ozcelik H, Ejlertsen B, Nielsen FC, Jonson L, Andersen MK, Ding YC, Steele L, Foretova L, Teule A, Lazaro C, Brunet J, Pujana MA, Mai PL, Loud JT, Walsh C, Lester J, Orsulic S, Narod SA, Herzog J, Sand SR, Tognazzo S, Agata S, Vaszko T, Weaver J, Stavropoulou AV, Buys SS, Romero A, de la Hoya M, Aittomaki K, Muranen TA, Duran M, Chung WK, Lasa A, Dorfling CM, Miron A, BCFR, Benitez J, Senter L, Huo D, Chan SB, Sokolenko AP, Chiquette J, Tihomirova L, Friebel TM, Agnarsson BA, Lu KH, Lejbkowicz F, James PA, Hall P, Dunning AM, Tessier D, Cunningham J, Slager SL, Wang C, Hart S, Stevens K, Simard J, Pastinen T, Pankratz VS, Offit K, Easton DF, Chenevix-Trench G, Antoniou AC, CIMBA (2013) Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet 9(3):e1003212. doi:10.1371/journal.pgen.1003212
Levy-Lahad E, Friedman E (2007) Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer 96(1):11–15
Chapman DD (2007) Cancer genetics. Semin Oncol Nurs 23(1):2–9
Cybulski C, Carrot-Zhang J, Kluzniak W, Rivera B, Kashyap A, Wokolorczyk D, Giroux S, Nadaf J, Hamel N, Zhang S, Huzarski T, Gronwald J, Byrski T, Szwiec M, Jakubowska A, Rudnicka H, Lener M, Masojc B, Tonin PN, Rousseau F, Gorski B, Debniak T, Majewski J, Lubinski J, Foulkes WD, Narod SA, Akbari MR (2015) Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet 47(6):643–646. doi:10.1038/ng.3284
Li Y, Chen Z (2004) Molecular cloning and characterization of LCRG1 a novel gene localized to the tumor suppressor locus D17S800-D17S930. Cancer Lett 209(1):75–85
Ohbayashi T, Oikawa K, Iwata R, Kameta A, Evine K, Isobe T, Matsuda Y, Mimura J, Fujii-Kuriyama Y, Kuroda M, Mukai K (2001) Dioxin induces a novel nuclear factor, DIF-3, that is implicated in spermatogenesis. FEBS Lett 508(3):341–344
Zhang J, Wang Y, Zhou Y, Cao Z, Huang P, Lu B (2005) Yeast two-hybrid screens imply that GGNBP1, GGNBP2 and OAZ3 are potential interaction partners of testicular germ cell-specific protein GGN1. FEBS Lett 579(2):559–566
Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG (1999) Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev 13(24):3198–3208
Hu X, Lazar MA (1999) The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402(6757):93–96
Kuang SQ, Liao L, Zhang H, Lee AV, O’Malley BW, Xu J (2004) AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res 64(5):1875–1885
Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA, Rosen EM (2006) The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol 20(1):14–34
Gizard F, Robillard R, Gross B, Barbier O, Revillion F, Peyrat JP, Torpier G, Hum DW, Staels B (2006) TReP-132 is a novel progesterone receptor coactivator required for the inhibition of breast cancer cell growth and enhancement of differentiation by progesterone. Mol Cell Biol 26(20):7632–7644
Li YJ, Xie HL, Chen ZC, He CM (2001) Cloning and expression analysis of a laryngeal carcinoma related gene, LCRG1. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 33(3):315–319
Guan R, Wen XY, Wu J, Duan R, Cao H, Lam S, Hou D, Wang Y, Hu J, Chen Z (2012) Knockdown of ZNF403 inhibits cell proliferation and induces G2/M arrest by modulating cell-cycle mediators. Mol Cell Biochem 365(1–2):211–222. doi:10.1007/s11010-012-1262-6
Yin F, Liu L, Liu X, Li G, Zheng L, Li D, Wang Q, Zhang W, Li L (2014) Downregulation of tumor suppressor gene ribonuclease T2 and gametogenetin binding protein 2 is associated with drug resistance in ovarian cancer. Oncol Rep 32(1):362–372. doi:10.3892/or.2014.3175
Bronzert DA, Greene GL, Lippman ME (1985) Selection and characterization of a breast cancer cell line resistant to the antiestrogen LY 117018. Endocrinology 117(4):1409–1417. doi:10.1210/endo-117-4-1409
Pei XH, Bai F, Smith MD, Usary J, Fan C, Pai SY, Ho IC, Perou CM, Xiong Y (2009) CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell 15(5):389–401
Hongo A, Kuramoto H, Nakamura Y, Hasegawa K, Nakamura K, Kodama J, Hiramatsu Y (2003) Antitumor effects of a soluble insulin-like growth factor I receptor in human ovarian cancer cells: advantage of recombinant protein administration in vivo. Cancer Res 63(22):7834–7839
Paliwal S, Kovi RC, Nath B, Chen YW, Lewis BC, Grossman SR (2007) The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res 67(19):9322–9329
Hu Y, Xu K, Yague E (2015) Mir-218 targets survivin and regulates resistance to chemotherapeutics in breast cancer. Breast Cancer Res Treat 151(2):269–280. doi:10.1007/s10549-015-3372-9
Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE (1988) Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 55(4):619–625
Holliday DL, Speirs V (2011) Choosing the right cell line for breast cancer research. Breast Cancer Res 13(4):215. doi:10.1186/bcr2889
Singhal H, Guo L, Bradlow HL, Mittelman A, Tiwari RK (1999) Endocrine characteristics of human breast epithelial cells, MCF-10F. Horm Res 52(4):171–177. doi:10.1159/000023457
Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, Tang P (2010) The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer (Auckl) 4:35–41
Chen Y, Chen C, Yang B, Xu Q, Wu F, Liu F, Ye X, Meng X, Mougin B, Liu G, Shen Z, Shao Z, Wu J (2011) Estrogen receptor-related genes as an important panel of predictors for breast cancer response to neoadjuvant chemotherapy. Cancer Lett 302(1):63–68. doi:10.1016/j.canlet.2010.12.014
Yamamoto-Ibusuki M, Arnedos M, Andre F (2015) Targeted therapies for ER+/HER2− metastatic breast cancer. BMC Med 13:137. doi:10.1186/s12916-015-0369-5
Ciarloni L, Mallepell S, Brisken C (2007) Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci USA 104(13):5455–5460
Zhang X, Xiao Z, Chen Z, Li C, Li J, Yanhui Y, Yang F, Yang Y, Oyang Y (2006) Comparative proteomics analysis of the proteins associated with laryngeal carcinoma-related gene 1. Laryngoscope 116(2):224–230
Duan CJ, Jiang TB, Li C (2008) Screening the effective target sequences of laryngeal carcinoma related gene LCRG1. Zhong Nan Da Xue Xue Bao Yi Xue Ban 33(6):468–475
Medina D (2005) Mammary developmental fate and breast cancer risk. Endocr Relat Cancer 12(3):483–495
Katzenellenbogen BS, Katzenellenbogen JA (2000) Estrogen receptor transcription and transactivation: estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res 2(5):335–344
Deroo BJ, Korach KS (2006) Estrogen receptors and human disease. J Clin Invest 116(3):561–570
Weihua Z, Andersson S, Cheng G, Simpson ER, Warner M, Gustafsson JA (2003) Update on estrogen signaling. FEBS Lett 546(1):17–24
Smith CL, O’Malley BW (2004) Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 25(1):45–71
McKenna NJ, Evans RM, O’Malley BW (2014) Nuclear receptor signaling: a home for nuclear receptor and coregulator signaling research. Nucl Recept Signal 12:e006. doi:10.1621/nrs.12006
Wysokinski D, Blasiak J, Pawlowska E (2015) Role of RUNX2 in breast carcinogenesis. Int J Mol Sci 16(9):20969–20993. doi:10.3390/ijms160920969
Heery DM, Kalkhoven E, Hoare S, Parker MG (1997) A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387(6634):733–736. doi:10.1038/42750
Plevin MJ, Mills MM, Ikura M (2005) The LxxLL motif: a multifunctional binding sequence in transcriptional regulation. Trends Biochem Sci 30(2):66–69. doi:10.1016/j.tibs.2004.12.001
Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, Raffoul W, Delaloye JF, Treboux A, Fiche M, Vilo J, Ayyanan A, Brisken C (2016) A preclinical model for eralpha-positive breast cancer points to the epithelial microenvironment as determinant of luminal phenotype and hormone response. Cancer Cell 29(3):407–422. doi:10.1016/j.ccell.2016.02.002
Acknowledgments
We thank Drs. Ray Wu, John Lydon, and Bert W. O’Malley for kindly providing human estrogen receptor expression vectors, ERE-luciferase plasmids, mammary RNA from normal and Wnt1 transgenic mice. We also thank Dr. Robert Clarke for graciously providing LCC9 and LY2 cells.
Funding
This work was supported in part by Grants R01-HD057501 (ZM Lei), 5P20RR017702-10, and 8P20 GM103453-10, project I (ZJ Lan) from the National Institutes of Health, USA, University of Louisville School of Medicine (CM Klinge), and 12ZCDZSY15700 (J Zhang) from Tianjin municipal Major Scientific and Technological Special Project for Significant Anticancer Development and Chinese National Natural Sciences Foundation 81402480 (YH Hu), China.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Zi-Jian Lan and YunHui Hu have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Lan, ZJ., Hu, Y., Zhang, S. et al. GGNBP2 acts as a tumor suppressor by inhibiting estrogen receptor α activity in breast cancer cells. Breast Cancer Res Treat 158, 263–276 (2016). https://doi.org/10.1007/s10549-016-3880-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-016-3880-2