
Abstract
Recurrence and death in a significant number of patients with ERα-positive breast cancer occurs 10–20 years after diagnosis. Prognostic markers for late events have been more elusive. TFAP2C (AP2γ) regulates the expression of ERα, the ERα pioneer factors FOXA1 and GATA3, and controls ERα-dependent transcription. The purpose of this investigation is to determine the long-term prognostic value of TFAP2C. A tissue microarray (TMA) consisting of breast tumors from 451 patients with median follow-up time of 10.3 years was created and tested for the expression of TFAP2C by immunohistochemistry. Wilcoxon Rank-Sum and Kruskal–Wallis tests were used to determine if TFAP2C H-scores correlate with other tumor markers. Cox proportional hazards regression models were used to determine whether TFAP2C H-scores and other tumor markers were related to overall and disease-free survival in univariate and multivariable models. TFPAC2 overexpression did not impact overall survival during the first 10 years after diagnosis, but was associated with a shorter survival after 10 years (HR 3.40, 95 % CI 1.58, 7.30; p value = 0.002). This late divergence persisted in ER-positive (HR 2.86, 95 % CI 1.29, 6.36; p value = 0.01) and endocrine therapy-positive subgroups (HR 4.19, 95 % CI 1.72, 10.23; p value = 0.002). For the ER+ and endocrine therapy subgroup, the HR was 3.82 (95 % CI 1.53, 9.50; p value = 0.004). TFAP2C H-scores were not correlated with other tumor markers or related to disease-free survival. In this hypothesis-generating study, we show that higher TFAP2C scores correlate with poor overall survival after 10 years of diagnosis in ERα-positive and endocrine therapy-treated subgroups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancers that express estrogen receptor alpha (ERα) are dependent on estrogen (E2) for growth and are responsive to anti-estrogen treatment [1]. The E2:ERα signaling network plays a role in breast cancer initiation and proliferation; however, that same network potentially prevents metastatic progression [2, 3]. 30–40 % of patients develop resistance to anti-estrogen treatment and these resistant tumors preferentially metastasize to bone [4, 5].
RNA-based intrinsic subtype classification and genome aberration-based integrative cluster analysis have subclassified ERα-positive breast cancers into multiple subtypes each with differing outcome [6, 7]. Thus, ERα-mediated signaling is not uniform across all ERα-positive breast cancers and tumor-specific genomic aberrations collude with ERα to confer diversity to ERα signaling and to overcome the effects of anti-estrogens. Recent studies have demonstrated that ERα itself undergoes mutations in metastatic lesions and these mutants are hypersensitive to E2 and/or resist the actions of ERα antagonists [8–10]. Apart from mutation in ERα, which is observed in 15–20 % of metastatic breast cancers [11], very limited data are available on the other genomic aberrations that collaborate with ERα to confer resistance to anti-estrogens.
Three distinct classes of signaling proteins control the function of ERα and aberrations in these proteins may provide an additional avenue for tumors to escape the actions of anti-estrogen. (1) Pioneer factors such as FOXA1, GATA3, TLE family members, and PBX1 enable the recruitment of ERα to chromatin [12, 13]. Transcriptional signatures that are dependent on ERα and specific pioneer factors identify tumors with differing response to endocrine therapy. For example, we, amongst others, have shown that elevated FOXA1 expression correlates with better response to anti-estrogen treatment [14–16]. However, there is some evidence for deleterious effects of FOXA1 if expressed in metastatic lesions and/or in tumors that have acquired anti-estrogen resistance [17, 18]. ERα-PBX1-dependent signature is associated with poor outcome [19]. (2) Co-activators and co-repressors. Among them, overexpression of AIB1 is linked to anti-estrogen resistance and metastatic progression in a subset of breast cancer [20]. (3) Kinases that phosphorylate ERα, which include AKT/PKB, ERK, SRC, PLK, and PKA [21]. However, elevated activity of these kinases is not always associated with poor outcome; elevated nuclear phospho-AKT(S473) correlates with better outcome in ERα-positive breast cancer [22]. There are conflicting reports on the role of ERα:PLK1 axis in anti-estrogen response and outcome [23, 24].
The focus of this study was on the pioneer factor TFAP2C, which regulates ERα activity at multiple levels. First, TFAP2C is required for the expression of ERα and its pioneer factors FOXA1 and GATA3 [25, 26]. Second, it is an E2-inducible gene [27]. Third, it is required for long-distance chromatin interaction and ERα-regulated gene expression including the genes that characterize ERα-positive breast cancer [28]. Regulatory regions of genes that are dependent on chromatin looping for expression are typically enriched for binding sites for ERα, FOXA1, and TFAP2C. Fourth, it controls luminal cell-specific unliganded ERα cistrome [29]. Fifth, it governs luminal epithelial phenotype of the breast by suppressing epithelial to mesenchymal transition [25]. ERα is expressed predominantly in luminal cells. Thus, TFAP2C is an integral part of the ERα:E2-mediated signaling in luminal breast epithelial cells.
Prognostic value of TFAP2C in breast cancer remains controversial. By analyzing 75 breast tumor tissues, Gee et al. reported poor outcome and anti-estrogen resistance in patients with TFAP2C overexpressing tumors [30]. Similar analysis of 54 tamoxifen-sensitive and 38 tamoxifen-resistant tumors revealed a role of elevated TFAP2C in tamoxifen resistance [31]. Friedrichs et al. reported no prognostic value for TFAP2C by analyzing 70 cases [32]. In animal models, TFAP2C has been shown to reduce ERBB2-mediated mammary tumor incidence but promote tumor progression [33]. Given the conflicting data, we sought to explore the prognostic value of TFAP2C in a large and well-annotated tissue microarray with long-term follow-up. We were particularly interested in the impact of TFAP2C on outcome 10 or more years after diagnosis given the paucity of biomarkers that can be used for predicting late recurrence and death [34, 35].
Materials and methods
REMARK guidelines for tumor biomarker reporting studies were followed in this study [36].
Patients, specimen characteristics and TMA construction
The TMA is composed of duplicate cores from 460 cases of breast cancer in 453 patients. Patients with duplicate cases, seven instances total, include two women with multiple primaries, four with lumpectomy and mastectomy tissue, and one with bilateral/synchronous disease. These samples were obtained from archival cases at Indiana University Health (University and Methodist Hospitals) between 1989 and 2003. Patient medical records were reviewed for demographics, pathology, treatment, and recurrence/follow-up information and are described in Table 1. For this study, we excluded males (n = 2). The Indiana University IRB approved this project.
Assay methods/immunostaining of TMA
Antibody against TFAP2C was purchased from Epitomics-Abcam (Cat#EP2692Y, Eugene, USA). The slides were deparaffinized in xylene and rehydrated through graded alcohols to water. Antigen retrieval was performed by immersing the slides in DAKO’s EnVision FLEX low pH target retrieval solution for 20 min. @ 100 °C., cooling to 85° and placed in Dako wash buffer. A 1:250 dilution of the primary antibody was added to the slides and incubated 20 min at room temperature. Following washing in TBST, visual detection was performed with DAKO Envison Plus Polymer Link and DAB chromogen (DAKO). The slides were washed and cover slipped. Control sections were treated with an isotype control using the same concentration as primary antibody to verify the staining specificity.
Whole slide digital imaging
The Aperio whole slide digital imaging system was used for imaging. The Aperio Scan Scope CS system was used (360 Park Center Drive, Vista, CA 92081, USA). The system imaged all slides at ×20. The scan time ranged from 1½ min to a maximum time of 2.25 min. The whole images were housed and stored in their Spectrum software system and images were shot from the whole slides.
Automatic image quantitation
The positive pixel count algorithm was used to quantify the amount of a specific stain present in a scanned slide image. A range of color (range of hues and saturation) and three intensity ranges (weak, positive, and strong) were masked and evaluated. The algorithm counted the number and intensity‐sum in each intensity range, along with three additional quantities: average intensity, ratio of strong/total number, and average intensity of weak positive pixels.
The algorithm had a set of default input parameters when first selected—these inputs have been pre‐configured for Brown color quantification in the three intensity ranges (220–175, 175–100, and 100–0). Pixels, which were stained, but did not fall into the positive‐color specification, were considered negative stained pixels—these pixels were counted as well, so that the fraction of positive to total stained pixels was determined.
The algorithm was applied to an image using ImageScope TMA Lab. This program allowed us to cover each tissue core in the array, specify the input parameters, run the algorithm, and view the results. Using the Aperio Positive Pixel algorithm, only staining classified as “positive” or “strong positive” was used to calculate positivity; regions classified as “weak positive” were mostly cytoplasmic and background staining, and were not counted.
Statistical methods
REMARK guidelines were followed for reporting study results [37]. For the seven subjects with multiple tumor samples available, we included only the sample with the highest TFAP2C H-score. T tests, Chi-square tests, Fisher’s Exact tests, or log-rank tests were used to compare patient and tumor variables between those with TFAP2C H-scores and those without. Wilcoxon Rank-Sum and Kruskal–Wallis tests were used to determine if TFAP2C H-scores were correlated with other tumor markers. Cox proportional hazards regression models were used to determine whether TFAP2C H-scores and other variables were related to overall and disease-free survival either univariately or in multivariable models. In these analyses, TFAP2C H-scores were divided into low and high categories at the score of 15.9858 for overall survival (time from surgery to death or censoring) and a score of 12.952 for disease-free survival (time from surgery to first recurrence or censoring, excluding patients with M1 stage at surgery). These cutoff values were determined using the maximum Chi-square value for all score values between the 25th and 75th percentile [38]. TFAP2C high/low was included in all multivariable models. Other tumor markers were included in multivariable models only if significant using a α level of 0.05 in the univariate models and to preserve the overall sample size we included missing as a separate category for PR status, tumor grade, and tumor stage. To assess non-proportional hazards, the AIC criterion was used to compare non-nested models. Using this criterion, the cutoff for choosing Model 1 over Model 2 is that the AIC for Model 1 is lower than that of Model 2 by at least 2.0. Overall and disease-free survival curves were generated for a priori specified subgroups (by ER status and endocrine therapy use) using the Kaplan–Meier Method and compared using log-rank tests. Analyses were conducted using SAS Version 9.4. A α level of 5 % was used to determine statistical significance.
To further extend observations made at protein levels to mRNA levels, we determined prognostic relevance of TFAP2C on overall and metastatic-free survival in various publicly available gene expression array datasets using our recently developed prognostic database PROGgene V2 [39]. The cBioportal database was used to determine genomic aberration in TFAP2C in various cancers [40].
Results
Of the 451 subjects in the study, 385 (85 %) had TFAP2C values available. Clinical parameters of the subjects included in the study are summarized in Table 1 with subgrouping of TFAP2C-positive and TFAP2C-negative cases. There were no differences in the characteristics of the patients or their tumors based on whether or not TFAP2C values were available. Median (range) follow-up from time of surgery was 10.3 years (0.04–21.8) for those with TFAP2C values and 10.3 years (1.1–17.5) for those without (p = 0.2421).
In the TFAP2C-positive group, there were 126 recurrences (33 %) and 163 deaths (42 %) during follow-up. The large number of HER-2/neu Unknown/Not Done was due to HER-2/neu testing not being fully integrated into institutional standard of care until April of 2001. There were no Unknown/Not Done values on or after that date. Also of note, 62/284 (22 %) of TFAP2C-postive ER-positive subjects did not receive endocrine therapy. The majority of these women (n = 46, 74 %) were diagnosed prior to 2000 when ER-positive disease was not well understood and aromatase inhibitors were unproven [41]. Tamoxifen was the only available treatment and often not prescribed to elderly women, those with small T1 tumors, or with other contraindication. Additionally, for premenopausal women diagnosed prior to 1998, tamoxifen was incorrectly thought not to be efficacious [42]. Specific reasons for lack of endocrine therapy in ER-positive group in this study include: patient refusal (n = 3, 5 %); comorbid condition (n = 4, 6 %); T1 tumor (n = 17; 28 %) age 70+ with or without T1 tumor or comorbidity (n = 23, 37 %); weakly staining tumor (n = 4, 6 %); premenopausal (n = 4, 6 %); and unknown (n = 7, 11 %). Endocrine therapy was primarily adjuvant treatment; only 4 % had known metastatic disease.
TFAP2C staining pattern in breast cancer
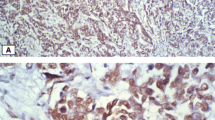
Representative TFAP2C immunostaining of breast tumors is shown in Fig. 1. The expression ranged from no expression to high expression and staining was restricted to nucleus. Interestingly, expression in normal breast was mosaic and staining intensity was similar to that of high expressing tumors. Thus, it is likely that tumors that express higher levels of TFAP2C have emerged from cells that naturally express higher levels of TFAP2C.

Immunostaining pattern of TFAP2C in breast cancers and normal breast
Correlation of TFAP2C H-score with other tumor markers
We generated TFAP2C H-score as described in the materials and methods and compared expression with ER, PR, HER-2/neu, Nodal stage and Grade. TFAP2C levels were not correlated with ER, PR, HER-2/neu, Nodal Stage, or Grade (Table 2).
Overall survival analysis
Univariate
In univariate analyses, variables significantly related to overall survival in the Cox proportional hazards regression models were PR status, tumor grade, tumor stage, and nodal stage (Table 3). PR−, higher tumor grade, higher tumor stage, and nodal stage-positive were correlated with lower survival. TFAP2C H-score was not related to overall survival (Fig. 2a, log-rank test p value 0.107); however, examination of the survival curve revealed that the survival curves for high and low TFAP2C were not parallel, indicating non-proportional hazards. The curves diverged at the 10-year mark. Cox models were compared that allowed for different hazard ratios for the time periods before or at 10 years and after 10 years verses proportional hazards using the AIC criterion, where it was found non-proportional hazard ratio provided a better fit. The results from these models (both univariate and multivariable adjusting for significant other tumor markers) indicated that the hazard ratio prior to or at 10 years was non-significant, but that the hazard ratio greater than 10 years was significantly different between the TFAP2C H-score categories. See Table 4 for multivariable model results. After 10 years, the hazard ratio as 3.40 (95 % CI 1.58, 7.30) when comparing High versus Low TFAP2C H-score categories.

Relationship between TFAP2C H-scores and overall survival of all patients or patients subdivided based on tumor characteristics and/or treatment. A blue line represents high TFAP2C H-score, whereas a red line represents low TFAP2C H-score
Subgroup analysis
We conducted subgroup analyses on overall survival using the ER-positive subgroup, endocrine therapy group, ER-positive on endocrine therapy, ER-negative, and ER+/PR+/HER2− (Fig. 2b–f). For ER-positive, endocrine therapy, and ER-positive on endocrine therapy, the results were similar to the overall results. Whether adjusted for covariates or not, hazard ratios were not significantly different from one prior to or at 10 years and were greater than one after 10 years. For ER-positive, HR 2.86 (95 % CI 1.29, 6.36). For endocrine therapy HR 4.19 (95 % CI 1.72, 10.23), and for ER-positive on endocrine therapy, HR 3.82 (95 % CI 1.53, 9.50). Results for ER-negative and ER+/PR+/HER2− were not significant.
Disease-free survival (DFS) analysis
Univariate
In univariate analyses, variables significantly related to disease-free survival in the Cox proportional hazards regression models were Tumor Grade, Tumor Stage, and Nodal Stage (Table S1). Higher Tumor Grade, Higher Tumor Stage, and Nodal Stage-positive were correlated with lower survival. TFAP2C H-score was not related to disease-free survival (log-rank test p value 0.2481, Fig. S1a).
Multivariable
In the multivariable analysis, Tumor Grade, Tumor Stage and Nodal Status were independently related to disease-free survival. Higher Tumor Grade and higher or unknown Tumor Stage were correlated with lower disease-free survival (Table S2).
Subgroup analysis
We conducted subgroup analyses on disease-free survival using the ER-positive subgroup, endocrine therapy group, ER-positive on endocrine therapy, ER-negative, and ER+/PR+/HER2− (Fig. S1b–f) but the results were not statistically significant (log-rank test p value 0.1097 for ER+, 0.1111 for those on endocrine therapy, 0.0707 for ER-positive on endocrine therapy, 0.8465 for ER-negative, and 0.1063 for ER+/PR+/HER2−).
Prognostic value of TFAP2C at mRNA level
Results of four mRNA analyses from public data are presented in Fig. 3a–d. In the gene expression array dataset described by Clarke et al., elevated expression of TFAP2C in grade 3 tumors was associated with poor overall survival [43]. Similarly, in Miller et al. dataset, elevated expression of TFAP2C in PR-negative tumors is associated with poor overall survival [44]. In the node-negative breast cancer dataset by Schmidt et al., TFAP2C overexpression was associated with poor metastasis-free survival and in the Loi et al. dataset, TFAP2C overexpression was associated with poor metastasis-free survival [45, 46].

Prognostic significance of TFAP2C mRNA in breast cancer. Public databases were used to classify breast tumors into TFAP2C-high and TFAP2C-low (divided at median) subgroups and then analyzed based on different tumor characteristics as indicated (a–d). e TFAP2C genome aberration in various cancers was determined using the cBioportal
cBioportal database analysis revealed TFAP2C amplification in a variety of cancers including 8 % of breast cancer [40]. Curiously, 28 % of primary breast cancers that were adapted grow as xenografts showed amplification (Fig. 3e). Thus, it appears that TFAP2C-overexpressing tumor cells are selected during tumor establishment/progression, at least under immunosuppressed condition.
Discussion
Although ERα-positive breast cancer is associated with better outcome compared with ERα-negative breast cancer, fear of recurrence is always in the minds of many patients. Since recurrence can occur even after 15 years of initial diagnosis, development of predictive biomarkers has proven difficult because of challenges in assimilating follow-up information for a considerably longer time. Having overcome this limitation, we present the role of TFAP2C in predicting recurrence and overall survival post-10 years of initial diagnosis. We saw no relationship between TFAP2C and disease recurrence. For overall survival, we saw no relationship between TFAP2C prior to or at 10 years (essentially negative results). After 10 years, the data suggests that higher TFAP2C was predictive of greater hazard of death. We do note that number of events is small in both High (n = 16 deaths) and Low (n = 12 deaths) TFAP2C H-Score groups after 10 years of diagnosis. In addition, because of lower number of events after 10 years, we were unable to divide our TMA into training and validation sets. For a simple log-rank test, 21 deaths total would be needed to have 80 % power to detect a hazard ratio of 3.4 using a two-sided test and 0.05 level of significance. Using a standard 50/50 split for the training and validation sets, we would only have 14 events per set. Therefore, results presented above should be considered “hypothesis generating” and need to be verified in a large TMA dataset with extensive follow-up as we have with our TMA. Few such TMAs have been published, which we hope to analyze in future [14, 47]. Inclusion of proliferation markers such as ki67 may allow us to determine whether TFAP2C expression is correlated with proliferation, and potentially luminal B phenotype, which is planned in future. However, the above limitation of the study is not due to quality of our TMA because in univariate and/or multivariate analysis, markers such as Tumor Grade, Tumor stage, and Nodal metastasis status were associated with overall survival and/or disease-free survival similar to many other published TMAs [16, 48, 49] (Tables 3, 4, S1, S2).
Amongst ERα pioneer factors examined for prognostic utility, PBX1 mRNA overexpression has been previously linked to poor outcome [19]. We had previously demonstrated better outcome in all and ERα-positive breast cancer patients with tumors that express higher levels of FOXA1 protein [16, 48]. Unlike the results of TFAP2C, differences in outcome between high and low FOXA1 expressers were evident as early as four-year post-diagnosis [16]. However, similar to TFAP2C, FOXA1 had no prognostic value in ERα-negative breast cancers. Thus, it is likely that prognostic utility of both FOXA1 and TFAP2C is linked to their pioneer factor activity on ERα. This is not unexpected based on intricate link in the expression/activity of ERα, TFAP2C, and FOXA1 with TFAP2C being the regulator of both ERα and FOXA1 [25]. However, downstream of this link, signaling by ERα:FOXA1 and ERα:TFAP2C may differ, which contributes to differences in tumor cell behavior. In this respect, TFAP2A, a closely related member of TFAP family, opposes the action of TFAP2C [50]. Balance between these two family members may ultimately determine the ability of TFAP2C in regulating ERα activity. An alternative possibility exits where TFAP2C and FOXA1 cooperate with each other in anti-estrogen-resistant breast cancers to afford distinct growth signaling pathway that does not require estrogen. Recent studies have demonstrated that the genomic activity of FOXA1 differs between tamoxifen-sensitive and tamoxifen-resistant breast cancer cells suggesting that tamoxifen-resistance mechanism involves switch in the activity rather than loss of FOXA1 expression [18, 51]. Since majority of TFAP2C:ERα regulated genes are also enriched for FOXA1 binding sites [28], it will be interesting to compare TFAP2C:FOXA1:ERα cistrome and transcriptome in anti-estrogen-sensitive and anti-estrogen-resistant cells.
Although chromosome 20q13.2 where TFAP2C is located is amplified in breast cancer [52], TFAP2C protein does not appear to be overexpressed in majority of breast cancer as we observed mosaic expression in normal breast and the expression levels in normal breast appear to be similar to that in cancers with highest TFAP2C H-score. Thus, it is likely that most of the cancers with elevated TFAP2C have originated from normal cells that naturally express higher levels of TFAP2C. Indeed, Oncomine database analysis did not reveal cancer-specific upregulation of TFAP2C except one study showing 1.2-fold increase in ductal carcinoma (data not shown). Different outcome observed in patients with tumors with higher and lower TFAP2C H-score or TFAP2C mRNA levels could be related to how ERα activity differs between TFAP2C-high and TFAP2C-low cells. To get insight into this possibility, we did coexpression analysis using Oncomine and cBioportal. Both databases revealed a network comprising TFAP2C, CITED1, CITED2, CITED4, and UBE2I. Among these genes, CITED1 is a co-activator of ERα activity and is required for mammary gland development [53, 54]. Thus, ERα:CITED1 transcriptional axis could differ in cells expressing higher levels of TFAP2C compared to cells with lower TFAP2C.
In vitro studies in cell lines have revealed a role for TFAP2C in inducing the expression of proto-oncogene RET independent of ERα [55]. TFAP2C has also been shown repress p21 in cooperation with cMyc oncogene [56]. Thus, tumors originating from cells with higher TFAP2C may be intrinsically more proliferative than tumors with lower TFAP2C. Tumors with higher proliferative rate usually respond initially to chemo or hormonal therapy but eventually recur. Future studies focusing on association between TFAP2C and proliferation markers such as ki67 may help establish a link between TFAP2C expression, proliferation, recurrence, and outcome.
References
Musgrove EA, Sutherland RL (2009) Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9:631–643
Torres-Arzayus MI, Zhao J, Bronson R, Brown M (2010) Estrogen-dependent and estrogen-independent mechanisms contribute to AIB1-mediated tumor formation. Cancer Res 70:4102–4111
Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS (2008) Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 456:663–666
Wang J, Jarrett J, Huang CC, Satcher RL Jr, Levenson AS (2007) Identification of estrogen-responsive genes involved in breast cancer metastases to the bone. Clin Exp Metastasis 24:411–422
Wei B, Wang J, Bourne P, Yang Q, Hicks D, Bu H, Tang P (2008) Bone metastasis is strongly associated with estrogen receptor-positive/progesterone receptor-negative breast carcinomas. Hum Pathol 39:1809–1815
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98:10869–10874
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486:346–352
Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H et al (2014) Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 20:1757–1767
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA et al (2013) ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45:1439–1445
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L et al (2013) Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45:1446–1451
Fuqua SA, Gu G, Rechoum Y (2014) Estrogen receptor (ER) alpha mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat 144:11–19
Nakshatri H, Badve S (2009) FOXA1 in breast cancer. Expert Rev Mol Med 11:e8
Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR et al (2005) Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122:33–43
Habashy HO, Powe DG, Rakha EA, Ball G, Paish C, Gee J, Nicholson RI, Ellis IO (2008) Forkhead-box A1 (FOXA1) expression in breast cancer and its prognostic significance. Eur J Cancer 44:1541–1551
Thorat MA, Marchio C, Morimiya A, Savage K, Nakshatri H, Reis-Filho JS, Badve S (2007) FOXA1 expression in breast cancer is associated with luminal subtype and good prognosis. J Clin Pathol 61:327–332
Badve S, Turbin D, Thorat MA, Morimiya A, Nielsen TO, Perou CM, Dunn S, Huntsman DG, Nakshatri H (2007) FOXA1 expression in breast cancer correlation with luminal subtype A and survival. Clin Cancer Res 13:4415–4421
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR et al (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481:389–393
Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS (2011) FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet 43:27–33
Magnani L, Ballantyne EB, Zhang X, Lupien M (2011) PBX1 genomic pioneer function drives ERalpha signaling underlying progression in breast cancer. PLoS Genet 7:e1002368
Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R (2003) Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 95:353–361
Anbalagan M, Rowan BG (2015) Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol. doi:10.1016/j.mce.2015.01.016
Badve S, Collins NR, Bhat-Nakshatri P, Turbin D, Leung S, Thorat M, Dunn SE, Geistlinger TR, Carroll JS, Brown M et al (2010) Subcellular localization of activated AKT in estrogen receptor- and progesterone receptor-expressing breast cancers: potential clinical implications. Am J Pathol 176:2139–2149
Wierer M, Verde G, Pisano P, Molina H, Font-Mateu J, Di Croce L, Beato M (2013) PLK1 signaling in breast cancer cells cooperates with estrogen receptor-dependent gene transcription. Cell Rep 3:2021–2032
Bhola NE, Jansen VM, Bafna S, Giltnane JM, Balko JM, Estrada MV, Meszoely I, Mayer I, Abramson V, Ye F et al (2015) Kinome-wide functional screen identifies role of PLK1 in hormone-independent, ER-positive breast cancer. Cancer Res 75:405–414
Cyr AR, Kulak MV, Park JM, Bogachek MV, Spanheimer PM, Woodfield GW, White-Baer LS, O’Malley YQ, Sugg SL, Olivier AK et al (2015) TFAP2C governs the luminal epithelial phenotype in mammary development and carcinogenesis. Oncogene 34:436–444
Woodfield GW, Chen Y, Bair TB, Domann FE, Weigel RJ (2010) Identification of primary gene targets of TFAP2C in hormone responsive breast carcinoma cells. Genes Chromosom Cancer 49:948–962
Orso F, Cottone E, Hasleton MD, Ibbitt JC, Sismondi P, Hurst HC, De Bortoli M (2004) Activator protein-2gamma (AP-2gamma) expression is specifically induced by oestrogens through binding of the oestrogen receptor to a canonical element within the 5′-untranslated region. Biochem J 377:429–438
Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, Yong EL, Sung WK, Cheung E (2011) AP-2gamma regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J 30:2569–2581
Caizzi L, Ferrero G, Cutrupi S, Cordero F, Ballare C, Miano V, Reineri S, Ricci L, Friard O, Testori A et al (2014) Genome-wide activity of unliganded estrogen receptor-alpha in breast cancer cells. Proc Natl Acad Sci USA 111:4892–4897
Gee JM, Eloranta JJ, Ibbitt JC, Robertson JF, Ellis IO, Williams T, Nicholson RI, Hurst HC (2009) Overexpression of TFAP2C in invasive breast cancer correlates with a poorer response to anti-hormone therapy and reduced patient survival. J Pathol 217:32–41
Guler G, Iliopoulos D, Guler N, Himmetoglu C, Hayran M, Huebner K (2007) Wwox and Ap2gamma expression levels predict tamoxifen response. Clin Cancer Res 13:6115–6121
Friedrichs N, Jager R, Paggen E, Rudlowski C, Merkelbach-Bruse S, Schorle H, Buettner R (2005) Distinct spatial expression patterns of AP-2alpha and AP-2gamma in non-neoplastic human breast and breast cancer. Mod Pathol 18:431–438
Jager R, Friedrichs N, Heim I, Buttner R, Schorle H (2005) Dual role of AP-2gamma in ErbB-2-induced mammary tumorigenesis. Breast Cancer Res Treat 90:273–280
Sgroi DC, Carney E, Zarrella E, Steffel L, Binns SN, Finkelstein DM, Szymonifka J, Bhan AK, Shepherd LE, Zhang Y et al (2013) Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 105:1036–1042
Bianchini G, Pusztai L, Karn T, Iwamoto T, Rody A, Kelly C, Muller V, Schmidt S, Qi Y, Holtrich U et al (2013) Proliferation and estrogen signaling can distinguish patients at risk for early versus late relapse among estrogen receptor positive breast cancers. Breast Cancer Res 15:R86
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM (2006) REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat 100:229–235
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM (2005) REporting recommendations for tumor MARKer prognostic studies (REMARK). Nat Clin Pract Urol 2:416–422
Zhou Q, Dhungana B (2012) A SAS® Macro for biomarker analysis using maximally selected Chi square statistics with application in oncology. PharmaSUG 2012:SP12
Goswami CP, Nakshatri H (2014) PROGgeneV2: enhancements on the existing database. BMC Cancer 14:970
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Bonneterre J, Thurlimann B, Robertson JF, Krzakowski M, Mauriac L, Koralewski P, Vergote I, Webster A, Steinberg M, von Euler M (2000) Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the Tamoxifen or Arimidex Randomized Group Efficacy and Tolerability study. J Clin Oncol 18:3748–3757
Early Breast Cancer Trialists’ Collaborative Group (1998) Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351:1451–1467
Clarke C, Madden SF, Doolan P, Aherne ST, Joyce H, O’Driscoll L, Gallagher WM, Hennessy BT, Moriarty M, Crown J et al (2013) Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis 34:2300–2308
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J (2005) An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 102:13550–13555
Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, Lehr HA, Hengstler JG, Kolbl H, Gehrmann M (2008) The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res 68:5405–5413
Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA et al (2007) Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol 25:1239–1246
Sarwar N, Kim JS, Jiang J, Peston D, Sinnett HD, Madden P, Gee JM, Nicholson RI, Lykkesfeldt AE, Shousha S et al (2006) Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr Relat Cancer 13:851–861
Mehta RJ, Jain RK, Leung S, Choo J, Nielsen T, Huntsman D, Nakshatri H, Badve S (2012) FOXA1 is an independent prognostic marker for ER-positive breast cancer. Breast Cancer Res Treat 131:881–890
Habashy HO, Rakha EA, Aleskandarany M, Ahmed MA, Green AR, Ellis IO, Powe DG (2011) FOXO3a nuclear localisation is associated with good prognosis in luminal-like breast cancer. Breast Cancer Res Treat 129:11–21
Bogachek MV, Chen Y, Kulak MV, Woodfield GW, Cyr AR, Park JM, Spanheimer PM, Li Y, Li T, Weigel RJ (2014) Sumoylation pathway is required to maintain the basal breast cancer subtype. Cancer Cell 25:748–761
Wright TM, Wardell SE, Jasper JS, Stice JP, Safi R, Nelson ER, McDonnell DP (2014) Delineation of a FOXA1/ERalpha/AGR2 regulatory loop that is dysregulated in endocrine therapy-resistant breast cancer. Mol Cancer Res 12:1829–1839
Nikolsky Y, Sviridov E, Yao J, Dosymbekov D, Ustyansky V, Kaznacheev V, Dezso Z, Mulvey L, Macconaill LE, Winckler W et al (2008) Genome-wide functional synergy between amplified and mutated genes in human breast cancer. Cancer Res 68:9532–9540
Yahata T, Shao W, Endoh H, Hur J, Coser KR, Sun H, Ueda Y, Kato S, Isselbacher KJ, Brown M, Shioda T (2001) Selective coactivation of estrogen-dependent transcription by CITED1 CBP/p300-binding protein. Genes Dev 15:2598–2612
Howlin J, McBryan J, Napoletano S, Lambe T, McArdle E, Shioda T, Martin F (2006) CITED1 homozygous null mice display aberrant pubertal mammary ductal morphogenesis. Oncogene 25:1532–1542
Spanheimer PM, Woodfield GW, Cyr AR, Kulak MV, White-Baer LS, Bair TB, Weigel RJ (2013) Expression of the RET proto-oncogene is regulated by TFAP2C in breast cancer independent of the estrogen receptor. Ann Surg Oncol 20:2204–2212
Wong PP, Miranda F, Chan KV, Berlato C, Hurst HC, Scibetta AG (2012) Histone demethylase KDM5B collaborates with TFAP2C and Myc to repress the cell cycle inhibitor p21(cip) (CDKN1A). Mol Cell Biol 32:1633–1644
Acknowledgments
We thank Janet Harlan and Jeanette Krohne for chart review, Lee Ann Baldridge for immunohistochemistry of TMA. IUPUI Signature center, IUSCC Breast Cancer Program, IUSCC Tissue Bank, and a grant from the Susan G. Komen for the Cure (SAC110025 to HN) supported this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Standards
The study was conducted in compliance with current laws of United States of America.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Perkins, S.M., Bales, C., Vladislav, T. et al. TFAP2C expression in breast cancer: correlation with overall survival beyond 10 years of initial diagnosis. Breast Cancer Res Treat 152, 519–531 (2015). https://doi.org/10.1007/s10549-015-3492-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3492-2