
Abstract
Multi-gene panels are used to identify genetic causes of hereditary breast and ovarian cancer (HBOC) in large patient cohorts. This study compares the diagnostic workflow in two centers and gives valuable insights into different next-generation sequencing (NGS) strategies. Moreover, we present data from 620 patients sequenced at both centers. Both sequencing centers are part of the German consortium for hereditary breast and ovarian cancer (GC-HBOC). All 620 patients included in this study were selected following standard BRCA1/2 testing guidelines. A set of 10 sequenced genes was analyzed per patient. Twelve samples were exchanged and sequenced at both centers. NGS results were highly concordant in 12 exchanged samples (205/206 variants = 99.51 %). One non-pathogenic variant was missed at center B due to a sequencing gap (no technical coverage). The custom enrichment at center B was optimized during this study; for example, the average number of missing bases was reduced by a factor of four (vers. 1: 1939.41, vers. 4: 506.01 bp). There were no sequencing gaps at center A, but four CCDS exons were not included in the enrichment. Pathogenic mutations were found in 12.10 % (75/620) of all patients: 4.84 % (30/620) in BRCA1, 4.35 % in BRCA2 (27/620), 0.97 % in CHEK2 (6/620), 0.65 % in ATM (4/620), 0.48 % in CDH1 (3/620), 0.32 % in PALB2 (2/620), 0.32 % in NBN (2/620), and 0.16 % in TP53 (1/620). NGS diagnostics for HBOC-related genes is robust, cost effective, and the method of choice for genetic testing in large cohorts. Adding 8 genes to standard BRCA1- and BRCA2-testing increased the mutation detection rate by one-third.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common cancer in women and over 10 % of all women will develop breast cancer during their life [1]. Twelve percent of women with breast cancer have at least one affected relative [2]. In these families, BRCA1 and BRCA2 were identified as high-risk genes for hereditary breast and ovarian cancer (HBOC) [3, 4]. The average lifetime risk for breast cancer in mutation carriers ranges from 50 to 80 % for BRCA1 and from 40 to 70 % for BRCA2 [5–8], respectively. The inheritance is autosomal dominant and the diagnosis of HBOC is not only relevant for the patient but also for other family members. The discovery of BRCA1 and BRCA2 led to the implementation of genetic testing in high-risk families and establishment of testing guidelines (www.nccn.org, www.awmf.org). Increased surveillance with specific screening protocols or prophylactic procedures is offered to mutation carriers. Today, more than 3500 genetic variants in BRCA1/2 are known (research.nhgri.nih.gov/bic). However, germline mutations in BRCA1 or BRCA2 account for only 20–40 % of high-risk families and over 60 % of the hereditary predisposition remains unexplained [9–12].
In search of additional causes for HBOC, several genes were identified during the last decade and classified as high-risk, intermediate-risk, or low-risk genes. Amongst the most important genes, in addition to BRCA1 and BRCA2, are CDH1, NBN, NF1, TP53, PTEN, STK11, ATM, BRIP1, CHEK2, PALB2, RAD50, RAD51C, and RAD51D [13–15]. Although the number of causative genes increased, the current gold standard in HBOC diagnostics is still Sanger sequencing of BRCA1 and BRCA2.
Next-generation sequencing (NGS) has evolved as a diagnostic tool that allows targeted sequencing of hundreds of genes and patients in parallel [16, 17]. With this approach, several clinical actionable genes can be targeted simultaneously. An increasing number of studies are published using NGS to screen large cohorts of patients [18]. These data also illustrate the variety of different study settings and sequencing set-ups. Here we compare results from two centers of the German consortium for hereditary breast and ovarian cancer (GC-HBOC) that offer diagnostic multi-gene testing for patients at risk. Both centers use different enrichment strategies and bioinformatic analysis. In addition, we present the mutational spectrum in 620 German HBOC patients that were sequenced at both centers.
Materials and methods
Patients
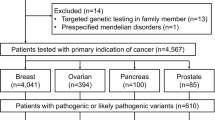
Informed consent was obtained from 620 patients with a high-risk family profile indicative for HBOC (for criteria s. Table 1). The GC-HBOC proposed a panel of 10 high- and intermediate-risk genes for analysis under research conditions to establish lifetime cancer risks and recommendations for future screening programs. These genes were selected and analyzed in all patients included in this study (BRCA1, BRCA2, RAD51C, RAD51D, PALB2, NBN, ATM, CDH1, TP53, CHEK2). Samples of 12 patients were blinded, exchanged, and sequenced at both centers.
Enrichment and sequencing
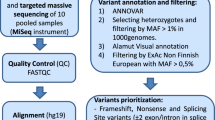
Center A implemented a commercial enrichment kit based on random enzymatic fragmentation, hybridization, and amplification (TruSight Cancer, Illumina, San Diego CA). This panel targets the coding exons of 94 genes and an additional set of 284 SNPs relevant in hereditary cancer syndromes. All samples were processed according to the manufacturer’s protocols. Center B used different versions of a custom enrichment based on fragmentation at defined restriction sites, hybridization, and amplification of the resulting fragments (HaloPlex, Agilent, Santa Clara CA). The selected genomic target region was designed using SureDesign (https://earray.chem.agilent.com/suredesign). All coding exons and adjacent intronic sequences ± 10 base pairs of 56–68 genes were included. The optimized design is available upon request. Both centers used a MiSeq (Illumina, San Diego) in paired-end mode to sequence the samples.
Bioinformatic analysis
The analysis was focused on all CCDS exons ± 2 base pairs: BRCA1—CCDS11453.1, CCDS11454.2, CCDS11455.2, CCDS11456.2, CCDS11459.2; BRCA2—CCDS9344.1; RAD51C—CCDS11611.1; RAD51D—CCDS11287.1, CCDS11288.1, CCDS45646.1; PALB2—CCDS32406.1; NBN—CCDS6249.1; ATM—CCDS31669.1; CDH1—CCDS10869.1; TP53—CCDS11118.1, CCDS45605.1, CCDS45606.1; CHEK2—CCDS13843.1, CCDS13844.1, CCDS33629.1, CCDS58798.1. The complete target region included 40 123 base pairs. Target regions that were not covered by a minimum depth of 20 reads were flagged as “low coverage nucleotides” and excluded from further analysis. Center A used a commercial software with standard parameters for mapping and variant calling (CLC Genomic Workbench, CLCbio, Denmark). The bioinformatics pipeline at center B included open-source tools for data analysis: stampy (vers. 1.0.23) combined with BWA (vers. 0.7.10) for mapping against hg19 [20, 21], samtools (vers. 1.1) for SAM/BAM-file handling, and variant calling [22]. All variants were left-aligned using GATK (vers. 3.2.2) [23], and own scripts were used for adapter trimming, quality control, and annotation of in-house variant frequencies. Annovar (vers. 21.06.2013) was used for annotation of variants [24]. This pipeline was designed to detect variants with a minimum allele frequency of 15 %. Both centers used IGV to optically validate sequencing results [25]. The Fisher's exact test was used for comparison of mutation rates in different genes (R v. 3.0.2). The R-package mada (v 0.5.4) was used without continuity correction for calculation of diagnostic sensitivity, specificity, and respective confidence intervals. Circos was used to visualize genetic data [26].
Interpretation of variants
Variants were categorized in a 5-class system (modified from Plon et al. [27]): pathogenic = variants with sufficient evidence for pathogenicity (e.g., variants resulting in premature truncation, functionally validated splicing mutations, gross genomic deletions); likely pathogenic = variants with strong evidence in favor of pathogenicity [e.g., variants at the canonical donor/acceptor sites (± 1 and 2), highly conserved rare variant (<1 %) within a known functional domain, expert review obligatory]; unknown significance = variants with insufficient or conflicting evidence regarding pathogenicity; all other variants were classified as benign (variants with very strong evidence against pathogenicity, e.g., mutation frequency in general population or subpopulation is too high) or likely benign (variants with strong evidence against pathogenicity, e.g., non-segregation with disease, silent variant with no predicted impact on splicing) were excluded from analysis. All pathogenic mutations were validated by Sanger sequencing.
Results
The aim of this study was the evaluation and comparison of NGS diagnostics at two sequencing centers. Both centers use different enrichment technologies and bioinformatic analysis strategies. Twelve samples were exchanged and analyzed at both centers in parallel. The overall turn-around time was 14 days per sample (sequencing: 8 days, bioinformatics: 3 days, medical report 2 days). Generally, test results should be available within 2–3 months. On average 17 variants were found per sample (min. 9, max. 24). With the support of an in-house database that collects variants and quality control values (which excluded frequent technical artifacts and variants with population frequencies >1 %), 0–1 variants per sample required further manual inspection and classification.
Custom enrichment—optimization
Center B used a custom enrichment that included the CCDS coding sequence and splice sites of all 10 genes to sequence 349 patients. It was optimized throughout this study to close sequencing gaps and increase the performance. The optimization included rearrangement and addition of hybridization probes (by multiplication of the same target region), increase of read lengths, and increase of sequencing output per sample. 22 samples were sequenced per MiSeq run and an average of 1.58 x106 reads per sample (min. 0.47 × 106 reads, max. 6.21 × 106 reads) was generated. The average amount of missing bases, i.e., bases covered by less than 20×, was reduced by a factor of four over time (s. Fig. 1): version 1—1939.41 bp (min. 623. bp, max. 4016 bp), version 2—817.37 bp (min. 328 bp, max. 2108 bp), version 3—605.85 bp (min. 300 bp, max. 2439 bp), and version 4—506.01 bp (min. 109 bp, max. 2026 bp). Although the number of bases with insufficient coverage could be reduced, not all sequencing gaps were completely closed within the target region. In addition, the variation of missing bases within the same enrichment version indicates that other confounders, e.g., sample quality, are also important for the number and size of missing regions.

Custom enrichment—optimization The number of missing bases within the 10 genes could be gradually reduced over different versions of the custom enrichment used at center B. Although the number of missing bases could be reduced by a factor of four, an average of 506.01 base pairs was not covered sufficiently in version 4. Legend a illustrates the performance of two different versions of the custom enrichment. The outer ring of the circos plot (brown) represents all 10 genes that were sequenced per patient. The inner ring shows all variants that were found as a scatter plot. The middle ring illustrates coverage data for all CCDS exons ± 2 base pairs and parts of an exon can be colored either green (covered well in >40 % of all samples) or red (all other regions).b gives a tabular overview of basic sequencing characteristics
Comparison of both centers—positive controls
Twelve samples were exchanged and sequenced at both centers. A total of 206 occurrences of 49 variants were found within all 10 genes. The overlap between both centers was 99.5 % (205/206 variants). All pathogenic mutations within the exchanged 12 samples were identified by both centers. Sequencing characteristics are illustrated in Fig. 2. A single variant in CHEK2 was not detected by center B because of insufficient read coverage (s. suppl. Fig. 1). In a diagnostic setting, this gap would have been closed by Sanger sequencing. Supplemental Fig. 1 also illustrates the differences between both enrichments: The enrichment strategy used at center B resulted in an amplicon-like dataset (fragments with the same chromosomal starting and end points), whereas center A generated reads that are randomly distributed over the target regions. Most common open-source tools require datasets with reads that have a random distribution over the target region by default. For this reason, the analysis of data at center B required modifications to standard software parameters (e.g., no strand bias filter, no random distribution of insert size; a complete list of all commands can be provided upon request). In summary, the sensitivity for center A was 100 % (CI 98.2–100.0 %) and 99.5 % (CI 97.3–99.9 %) for center B. The analytical specificity was estimated by the evaluation of 167 exons known to be homozygous wild type or carry polymorphisms and it was found to be 100 % (CI 97.8–100 %) for center A and B. Finally, the number of missing base pairs, i.e., base pairs that are not covered by at least 20 reads, is important since missing base pairs would have to be sequenced by a second independent method in a diagnostic setting. To compare the results from both centers, we used the complete CCDS regions of all 10 genes. A mean of 316.67 bp (min. 253 bp, max. 415 bp) was missing at center A. Most missing bases were not covered by the enrichment design: one exon in RAD51D, one exon in BRCA1, and two exons in TP53. These exons were identified to contain repetitive elements that are difficult to target by this type of enrichment. There were no additional gaps in the remaining genes. A mean of 653.83 bp (min. 208 bp, max. 1793 bp) was not covered sufficiently at center B. Missing bases were found in 12 exons of BRCA1, BRCA2, CHEK2, RAD51C, and ATM. Finally, custom enrichments provide flexibility in optimization of sequencing performance and selection of target regions. The total size of target regions has direct influence on sequencing costs. Center A generally sequences 12 samples per MiSeq run and center B 22.

Comparison of both centers—positive controls A total of 12 samples were sequenced and analyzed at both centers. Legend a gives an overview of the results. The overlap of both centers was 206/207 variants. One variant in CHEK2 was missed at center B since it was located within a sequencing gap. CCDS exons were used for data comparison, and an average of 316.67 base pairs was not covered sufficiently by center A, 653.83 base pairs by center B. Missing bases at center A were not part of the enrichment’s target region. Therefore, all targeted regions were covered by at least 20× (asterisk). Within all 12 samples, no pathogenic variant was missed at both centers. a illustrates the sequencing results from both centers. The outer ring of the circos plot (brown) represents all 10 genes that were sequenced per patient. The inner ring shows all variants that were found as a scatter plot. The middle ring illustrates coverage data for all CCDS exons ± 2 basepairs. Parts of each exon can be colored either green (covered well in >40 % of all samples) or red (all other regions). Part B gives an overview of basic sequencing characteristics

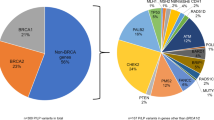
Frequency of mutations within all core genes. a shows the frequency of mutations identified in our set of 620 patients and compares the results to data published by Castera et al. [39] (mutations and potential mutations). The pie chart in (b) illustrates the distribution of mutations
Mutational landscape—620 patients
63 different mutations that were classified as likely pathogenic or pathogenic with 75 occurrences in different patients were found in all 10 genes analyzed in this study (s. Fig. 3 and supplement). 57 patients carried a pathogenic mutation in BRCA1 and BRCA2 (9.19 %, 57/620). Overall, we were able to identify a genetic cause for breast cancer in 12.10 % (75/620) of all families (s. Fig. 3). Mutations in BRCA1 were most frequent (30 pts), followed by BRCA2 (27 pts), CHEK2 (6 pts), ATM (4 pts), CDH1 (3 pts), NBN (2 pts), PALB2 (2 pts), and TP53 (1 pt). No mutations were found in RAD51C and RAD51D. Standard sequencing of BRCA1 or BRCA2 would have identified HBOC causing mutations in 9.19 %, whereas NGS and the addition of 8 genes increased the mutation detection rate by 2.91 % and consequently answered the causal event for an additional one-third of HBOC patients. A list of pathogenic mutations can be found in the supplement of this article. No significant differences in mutation detection rates were found between both centers.
Discussion
NGS has become the most important technology for DNA analysis with comparable outcome in sensitivity and specificity to Sanger sequencing [28]. Several studies have confirmed the suitability of NGS in HBOC diagnostics and have recently been reviewed by Kurian et al. [18]. Yet, a large variety of different methods for enrichment, sequencing, and bioinformatics analysis are in use. For example, Cybulski et al. [29] used exome sequencing that offers the highest flexibility in target selection but still seems to be too expensive and not sensitive enough for routine diagnostics. In contrast, other studies focused on solely sequencing of BRCA1 and BRCA2, probably missing a relevant fraction of pathogenic mutations in other HBOC genes including TP53 and PTEN [30, 31]. All 10 genes that were included in this study are known to be high- or intermediate-risk genes for HBOC [15, 32, 33]. Most current studies use multi-gene panels of different compositions that predominantly include genes of the DNA repair pathways [34–37]. These panels represent a compromise between costs, sensitivity, and flexibility. Custom enrichments offer the opportunity for manual optimization and flexible adaption of target regions. The recommended setup for a medium-sized diagnostic laboratory (400 samples/year) should include one to two technicians, one bioinformatician, one biologist, and two (desktop) sequencers.
Comparison of two sequencing centers
This study compares data from two GC-HBOC centers that offer NGS for HBOC diagnostics. The results confirmed the high reproducibility of NGS: 99.5 % of all variants (205/206) were found by both centers within 12 exchanged samples. Center B missed one (non-pathogenic) variant in CHEK2 located in a low-covered region. These low-covered regions are relevant in a diagnostic setting and have to be filled by a second independent method. We defined sequencing gaps as target regions covered by less than 20 reads per base. This is concordant to the literature, where the required sequencing depth ranges from 20 to 50 reads [35, 37]. With the recent version of the custom enrichment, a mean of 653.83 nucleotides (1.63 %) was covered by less than 20 reads. Recurrent low-coverage regions were found in 5 genes and included 12 different exons. Arvai et al. [38] used the same enrichment in combination with a different sequencing device and reported 3.55 % of their target region that was covered by less than 20 reads. The target region at center A was sequenced without any sequencing gaps. Yet, four CCDS exons in three different genes (BRCA1, RAD51D, TP53) were missing in the design as they contained repetitive elements and were therefore not targetable with this type of enrichment. For this reason, a mean of 316.67 nucleotides per sample within the four exons was not covered sufficiently (0.79 %). The enrichment system used at center A seemed to be more robust as the number of missing bases was very stable over all patients: Standard deviation at center A was 45.67 bp, at center B 444.42 bp. The distribution of low-coverage regions is important, since cost and expenditure of time increase with the number of exons that have to be reanalyzed with a second independent method (e.g., Sanger sequencing). Some reports confirm that low-coverage regions within the target regions may occur, although additional experiments are not always required [30, 35, 38]. These differences may be attributable to the definition of the target region and experimental setup. For this study, we used CCDS as reference sequence for comparative analysis.
Mutational landscape
Pathogenic BRCA1 and BRCA2 mutations were found in 9.19 % of our patients. The data presented in this study are comparable to a previous report that included patients from high-risk families (s. Fig. 3) [39]. This study reported a BRCA1/2 mutation detection rate of 10.8 % [39]. The addition of 8 other genes to standard BRCA1/2 diagnostics added 2.91 % of solved cases. This corresponds to a mutation detection rate of 3.20 % in BRCA1/2-negative patients (18/563). Previous reports found pathogenic mutations in BRCA1/2-negative patients in 6.45–11.40 % [34, 36, 39]. In this study, no mutations were found in RAD51C and RAD51D what may be due to patient selection criteria or cohort size. The data presented in this study are comparable to a previous report that included patients from high-risk families (s. Fig. 3) [39]. Patients with pathogenic or likely pathogenic mutations are offered increased surveillance or preventive surgery. With an increasing number of genes, variants are found that cannot be further classified today. The percentage of variants of unknown significance (VUS) was 33.33 % at center A and 27.59 % at center B. The reported fractions of VUSs are difficult to compare between different studies and depend on the target regions. Several groups reported overall VUS rates of 19–88 % [34, 36, 40]. With the application of NGS in standard diagnostics, the fraction of VUSs will decrease in the near future.
Conclusion
NGS has advantages over conventional Sanger sequencing when it comes to costs, throughput and turn-around time allowing improved diagnostics by analyzing a higher number of susceptibility genes and thus increasing the diagnostic yield. The selection and optimization of enrichment methods are mandatory as low-coverage regions need to be filled in by a second method to complete the analysis in a diagnostic setting. In this study, multi-gene panels helped to identify pathogenic mutations in additional 2.91 % of families. Surveillance protocols for mutation carriers within the ten genes sequenced in this study will be needed in the near future. Despite the recent efforts, the underlying hereditary cause in more than 80 % of all families admitted to genetic testing is still not identified. We will have to identify additional genes that contribute to HBOC while moving to a polygenic HBOC model. NGS is currently the only method that is able to cover large multi-gene panels in a high-throughput setting.
References
Ferlay J, Parkin DM, Steliarova-Foucher E (2010) Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer 46(4):765–781. doi:10.1016/j.ejca.2009.12.014
Collaborative Group on Hormonal Factors in Breast C (2001) Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 358(9291):1389–1399. doi:10.1016/S0140-6736(01)06524-2
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266(5182):66–71
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378(6559):789–792. doi:10.1038/378789a0
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, Eccles DM, Tang N, Olah E, Anton-Culver H, Warner E, Lubinski J, Gronwald J, Gorski B, Tulinius H, Thorlacius S, Eerola H, Nevanlinna H, Syrjakoski K, Kallioniemi OP, Thompson D, Evans C, Peto J, Lalloo F, Evans DG, Easton DF (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72(5):1117–1130. doi:10.1086/375033
Chen S, Iversen ES, Friebel T, Finkelstein D, Weber BL, Eisen A, Peterson LE, Schildkraut JM, Isaacs C, Peshkin BN, Corio C, Leondaridis L, Tomlinson G, Dutson D, Kerber R, Amos CI, Strong LC, Berry DA, Euhus DM, Parmigiani G (2006) Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol 24(6):863–871. doi:10.1200/JCO.2005.03.6772
Chen S, Parmigiani G (2007) Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 25(11):1329–1333. doi:10.1200/JCO.2006.09.1066
Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, Tang J, Li S, Zhang S, Shaw PA, Narod SA (2006) Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst 98(23):1694–1706. doi:10.1093/jnci/djj465
Wooster R, Weber BL (2003) Breast and ovarian cancer. The New Engl J Med 348(23):2339–2347. doi:10.1056/NEJMra012284
Nathanson KL, Wooster R, Weber BL (2001) Breast cancer genetics: what we know and what we need. Nat Med 7(5):552–556. doi:10.1038/87876
Meindl A, German Consortium for Hereditary B, Ovarian C (2002) Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int J Cancer 97(4):472–480
Turnbull C, Rahman N (2008) Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet 9:321–345. doi:10.1146/annurev.genom.9.081307.164339
Apostolou P, Fostira F (2013) Hereditary breast cancer: the era of new susceptibility genes. BioMed Res Int 2013:747318. doi:10.1155/2013/747318
Bradbury AR, Olopade OI (2007) Genetic susceptibility to breast cancer. Rev Endocr Metabol Disord 8(3):255–267. doi:10.1007/s11154-007-9038-0
Ripperger T, Gadzicki D, Meindl A, Schlegelberger B (2009) Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur J Hum Genet 17(6):722–731. doi:10.1038/ejhg.2008.212
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11(1):31–46. doi:10.1038/nrg2626
Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ (2010) Target-enrichment strategies for next-generation sequencing. Nat Methods 7(2):111–118. doi:10.1038/nmeth.1419
Kurian AW, Kingham KE, Ford JM (2015) Next-generation sequencing for hereditary breast and gynecologic cancer risk assessment. Curr Opin Obstet Gynecol 27(1):23–33. doi:10.1097/GCO.0000000000000141
Wockel A, Kreienberg R (2008) First revision of the German S3 guideline ‘diagnosis, Therapy, and follow-up of breast cancer’. Breast care 3(2):82–86. doi:10.1159/000127509
Lunter G, Goodson M (2011) Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21(6):936–939. doi:10.1101/gr.111120.110
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. doi:10.1093/bioinformatics/btp698
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. doi:10.1093/bioinformatics/btp352
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. doi:10.1038/ng.806
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. doi:10.1093/nar/gkq603
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29(1):24–26. doi:10.1038/nbt.1754
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645. doi:10.1101/gr.092759.109
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV, Group IUGVW (2008) Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29(11):1282–1291. doi:10.1002/humu.20880
Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van den Berg MP, van Spaendonck-Zwarts KY, van Tintelen JP, Sijmons RH, Jongbloed JD, Sinke RJ (2013) Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat 34(7):1035–1042. doi:10.1002/humu.22332
Cybulski C, Lubinski J, Wokolorczyk D, Kuzniak W, Kashyap A, Sopik V, Huzarski T, Gronwald J, Byrski T, Szwiec M, Jakubowska A, Gorski B, Debniak T, Narod SA, Akbari MR (2014) Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin Genet. doi:10.1111/cge.12524
Trujillano D, Weiss ME, Schneider J, Koster J, Papachristos EB, Saviouk V, Zakharkina T, Nahavandi N, Kovacevic L, Rolfs A (2015) Next-generation sequencing of the BRCA1 and BRCA2 genes for the genetic diagnostics of hereditary breast and/or ovarian cancer. J Mol Diagn 17(2):162–170. doi:10.1016/j.jmoldx.2014.11.004
Feliubadalo L, Lopez-Doriga A, Castellsague E, del Valle J, Menendez M, Tornero E, Montes E, Cuesta R, Gomez C, Campos O, Pineda M, Gonzalez S, Moreno V, Brunet J, Blanco I, Serra E, Capella G, Lazaro C (2013) Next-generation sequencing meets genetic diagnostics: development of a comprehensive workflow for the analysis of BRCA1 and BRCA2 genes. Eur J Hum Genet 21(8):864–870. doi:10.1038/ejhg.2012.270
Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K, Deissler H, Engel C, Muller-Myhsok B, Neveling K, Kiechle M, Mathew CG, Schindler D, Schmutzler RK, Hanenberg H (2010) Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 42(5):410–414. doi:10.1038/ng.569
Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, Bowden G, Kalmyrzaev B, Warren-Perry M, Snape K, Adlard JW, Barwell J, Berg J, Brady AF, Brewer C, Brice G, Chapman C, Cook J, Davidson R, Donaldson A, Douglas F, Greenhalgh L, Henderson A, Izatt L, Kumar A, Lalloo F, Miedzybrodzka Z, Morrison PJ, Paterson J, Porteous M, Rogers MT, Shanley S, Walker L, Breast Cancer Susceptibility C, Eccles D, Evans DG, Renwick A, Seal S, Lord CJ, Ashworth A, Reis-Filho JS, Antoniou AC, Rahman N (2011) Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet 43(9):879–882. doi:10.1038/ng.893
Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, Rathbun K, Stopfer JE, Zhu J, Bradbury AR, Simon MS, DeMichele A, Domchek SM, Nathanson KL (2014) Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med. doi:10.1038/gim.2014.176
Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC (2010) Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA 107(28):12629–12633. doi:10.1073/pnas.1007983107
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM (2014) Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 32(19):2001–2009. doi:10.1200/JCO.2013.53.6607
Chong HK, Wang T, Lu HM, Seidler S, Lu H, Keiles S, Chao EC, Stuenkel AJ, Li X, Elliott AM (2014) The validation and clinical implementation of BRCAplus: a comprehensive high-risk breast cancer diagnostic assay. PLoS ONE 9(5):e97408. doi:10.1371/journal.pone.0097408
Arvai K, Horvath P, Balla B, Tokes AM, Tobias B, Takacs I, Nagy Z, Lakatos P, Kosa JP (2014) Rapid and cost effective screening of breast and ovarian cancer genes using novel sequence capture method in clinical samples. Fam Cancer 13(4):583–589. doi:10.1007/s10689-014-9730-7
Castera L, Krieger S, Rousselin A, Legros A, Baumann JJ, Bruet O, Brault B, Fouillet R, Goardon N, Letac O, Baert-Desurmont S, Tinat J, Bera O, Dugast C, Berthet P, Polycarpe F, Layet V, Hardouin A, Frebourg T, Vaur D (2014) Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet 22(11):1305–1313. doi:10.1038/ejhg.2014.16
Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, Timms K, Garber JE, Herold C, Ellisen L, Krejdovsky J, DeLeonardis K, Sedgwick K, Soltis K, Roa B, Wenstrup RJ, Hartman AR (2015) Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 121(1):25–33. doi:10.1002/cncr.29010
Acknowledgments
The authors thank all patients and families for their participation in this study. This study was supported by the German Consortium for Hereditary Breast and Ovarian Cancer (GC-HBOC).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Schroeder, C., Faust, U., Sturm, M. et al. HBOC multi-gene panel testing: comparison of two sequencing centers. Breast Cancer Res Treat 152, 129–136 (2015). https://doi.org/10.1007/s10549-015-3429-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3429-9