
Abstract
The purpose of this study was to discover novel nuclear receptor targets in triple-negative breast cancer. Expression microarray, Western blot, qRT-PCR analyses, MTT growth assay, soft agar anchorage-independent growth assay, TRE reporter transactivation assay, and statistical analysis were performed in this study. We performed microarray analysis using 227 triple-negative breast tumors, and clustered the tumors into five groups according to their nuclear receptor expression. Thyroid hormone receptor beta (TRβ) was one of the most differentially expressed nuclear receptors in group 5 compared to other groups. TRβ low expressing patients were associated with poor outcome. We evaluated the role of TRβ in triple-negative breast cancer cell lines representing group 5 tumors. Knockdown of TRβ increased soft agar colony and reduced sensitivity to docetaxel and doxorubicin treatment. Docetaxel or doxorubicin long-term cultured cell lines also expressed decreased TRβ protein. Microarray analysis revealed cAMP/PKA signaling was the only KEGG pathways upregulated in TRβ knockdown cells. Inhibitors of cAMP or PKA, in combination with doxorubicin further enhanced cell apoptosis and restored sensitivity to chemotherapy. TRβ-specific agonists enhanced TRβ expression, and further sensitized cells to both docetaxel and doxorubicin. Sensitization was mediated by increased apoptosis with elevated cleaved PARP and caspase 3. TRβ represents a novel nuclear receptor target in triple-negative breast cancer; low TRβ levels were associated with enhanced resistance to both docetaxel and doxorubicin treatment. TRβ-specific agonists enhance chemosensitivity to these two agents. Mechanistically enhanced cAMP/PKA signaling was associated with TRβ’s effects on response to chemotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Progress has been made in treating patients with estrogen receptor alpha (ERα) or HER2-positive breast tumors [1–3]. However, triple-negative breast cancer (TNBC) (ERα-/progesterone receptor (PR)-/HER2-) is harder to treat as these tumors exhibit high proliferation rates, early recurrence, and decreased disease survival [4, 5]. TNBC is currently treated with chemotherapy due to the lack of ER and HER2 therapeutic targets [6–8]. Many TNBCs are sensitive to chemotherapy and treatment improves disease-free and overall survival [8, 9]. However, the problem of chemotherapy resistance is still a significant clinical problem [10, 11].
Taxanes, doxorubicin, and platinum compounds are used in TNBC [12–15]. Unfortunately there are few useful predictors of chemotherapeutic response [16, 17]. Since chemotherapy is often limited by toxicity, there is a need to explore biologic targets in this subtype. A recent genomic study identified gene pathways associated with chemotherapy sensitivity in breast cancer subtypes [18], and in the ER-negative subgroup, gene sets involved in elevated G-protein signaling, fatty acid synthesis, and cAMP-mediated signaling were associated with poor chemotherapeutic response [18]. Some TNBCs express elevated levels of the androgen receptor (AR), and one TN cell line has been shown to be androgen-responsive [19]. A molecular luminal AR (LAR) subgroup has also been defined in TNBC, and corresponding cell lines are responsive to AR antagonist treatment [20]. In a Phase II TNBC trial with AR-expressing tumors, AR targeted therapy demonstrated a 19 % clinical benefit rate [21]. Thus, with the success of targeting AR in TNBC, we hypothesized that other nuclear receptors (NRs) could be novel therapeutic targets in TNBC.
There are 48 members of the NR superfamily, and a recent study reported that many NR levels were lower in tumors compared to normal breast tissues [22]. It has been shown that thyroid hormone receptor beta (TRβ) acts as a tumor suppressor of invasion and metastasis in TNBC cell lines [23]. TRβ is a nuclear transcription factor that mediates the pleiotropic activities of thyroid hormones, T3 and T4, and influences basal oxygen consumption, cardiac contractility, and lipid metabolism [24–26]. Additionally, activation of TRs with T3 modulates aerobic glycolysis (the Warburg effect) in breast cancer cells [27]. TRβ levels, but not the closely related TRα isoform, were negatively associated with tumor grade [22]. TRβ promotes transcription in the presence of thyroid hormones, but inhibits transcription in absence of ligand [25]. Importantly specific TRβ agonists are available, and have been clinically successful in lowering triglyceride and cholesterol levels without inducing side effects in the heart which are mediated by TRα [28–30].
We recently reported RNA expression profiling of 198 TNBC and defined TNBC subtypes with distinct prognostic outcomes [31]. We used these array data to subtype TNBC using NR expression levels, and found that the TRs were overexpressed in one group, and elevated TRβ levels were associated with better clinical outcomes. The role of TRβ in breast cancer is not well studied. We discovered that low TRβ levels were associated with resistance to selected chemotherapeutic drugs, and treatment with a specific TRβ agonist restored chemo sensitivity in TNBC cells.
Results
Low levels of TRβ are associated with poor clinical outcomes and enhanced growth of TNBC
Treatment of TNBC suffers from the lack of clinical targets or known genetic drivers limiting gene-targeted approaches [32]. In order to explore whether NRs could be potential targets in TNBC, we used supervised hierarchical clustering to separate 227 TNBC into 5 potential subgroups (Supplemental Fig. 1a). NRs contributing to these clusters were scored using prediction analysis of microarray (PAM) across the clustered groups, and the average expression of the NRs determined (Fig. 1a). Group 5 represents 24 % of the total patients, and the most highly expressed receptors classifying this group are shown in Fig. 1b. Previous analysis of this microarray dataset using non-negative matrix factorization [31] classified AR-overexpressing tumors as the LAR group defined by Lehman [20]. LAR tumors are known to have abundant AR protein, are ER protein negative, but ER RNA positive. However, our NR-classified group 5 tumors do not overlap with Lehmann’s LAR group signature [20] (data not shown), but correspond to a group with high expression of several NRs, including AR, ER, COUP TF1, TRβ, and ERRs α/γ (Fig. 1b). To determine which TNBC cell line models could be used to explore the role of TRβ, we matched our available cells to the NR subgroups 1–5 using PAM classification [33], and examined levels of group 5 NRs in the cell lines using qRT-PCR and immunoblot analysis (Fig. 1c, d). Corresponding NR group 5 cell lines MDA-MB-453, HCC2185, and HCC202 showed higher levels of TRβ protein compared to NR group 3. Levels of AR were also higher in NR group 5 tumors, with no ER protein expressed as expected.

Low TRβ levels are associated with worse DFS and enhanced cell proliferation. a Heat map showing comparison of NR gene expression among five patient groups using Cluster 3.0 and Tree View. The comparison was calculated based on relative NR gene expression. Red or green color indicates relatively high or low expression, respectively, compared to other groups, and black indicates average expression. Some genes were recognized by multiple Affymetrix probes. b The most differentially overexpressed genes in group 5 are shown. c qRT-PCR was performed to evaluate the gene expression of TRβ, AR, and ERα in group 5 representative cell lines HCC202, MDA-MB-453, and HCC2185. 18S RNA was used as a loading control. Group 3 representative cell lines HCC38 and HCC1569 were used as negative controls. MCF-7 gene amplification cycles were used as the reference to calculate fold change. d TRβ, AR, and ERα protein expression were examined using Western blot analysis in group 5, group 3, and ER positive breast cancer cell lines. GAPDH was used as loading control. e Kaplan–Meier plot of TRβ using Sabatier et al. [34] (**p < 0.01). Data represent an average of 3 Affymetrix TRβ probesets, and 75th percentile was used as the cut-point to separate patients into two outcome groups. f HCC202 and MDA-MB-453 cells were stable transfected with empty vector (EV) or TRβ shRNA (SH) plasmid and TRβ expression was evaluated by Western blot and soft agar. Results represent the average ± SD of three experiments normalized to respective EV (*p < 0.05). g HCC2185 EV and SH cells were analyzed for TRβ expression using Western blot analysis and growth in MTT assays using GraphPad Prism 5 (***p < 0.001 SH cells growth curve compared to EV)
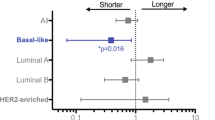
We first evaluated correlations between TRβ expression and patient survival using publicly available clinical data [34]. Patients with high TRβ mRNA levels were associated with longer disease-free survival (Fig. 1e). Since the majority of ER-negative patients in this dataset were treated with different chemotherapeutic regimens, we cannot differentiate TRβ effects on prognosis independent of treatment.
TRβ can affect invasion and metastasis in MDA-MB-486 ER-negative breast cancer cells [23]. To explore the role of TRβ on the growth of TNBC cells, we used shRNA knock down (KD) in three representative cell lines, and performed soft agar or MTT assays. HCC202 and MDA-MB-453 KD cells formed significantly more colonies compared with cells infected with empty vector (Fig. 1f, EV). MTT assays in HCC2185 confirmed these results (Fig. 1g). Inducible overexpression of TRβ in MDA-MB-453 cells reduced cell growth as expected and enhanced chemosensitivity (Supplemental Fig. 2). Our data suggest that TRβ could act as a tumor suppressor in TNBC cells.
TRβ KD enhances resistance to chemotherapy and blocks apoptosis of TNBC cells
To evaluate the effect of TRβ levels on response to chemotherapy regimens commonly used in TNBC, we determined IC50 values in TRβ KD and EV cells after treatment for 3 days with the taxane docetaxel (T), the anthracycline doxorubicin (D), or cisplatin (C). TRβ KD increased the IC50 vales for docetaxel and doxorubicin in both cell lines (Fig. 2a, b, respectively). IC50 values for docetaxel and doxorubicin were increased 5–6 fold and 4–7-fold, respectively, in HCC2185 and HCC202 cells. However, the IC50 for cisplatin did not change with TRβ KD. To test whether drug treatment might also affect TRβ expression, we cultured NR group 5 cell lines for 2 months in drug. TRβ protein levels decreased in all three cell lines with continuous docetaxel and doxorubicin treatments (Fig. 2c–e). In HCC202 and MDA-MB-453 cells, TRβ levels decreased in a dose-dependent manner. These data suggest that treatment itself can decrease TRβ levels, which could further contribute to resistance and low TRβ expression could be a predictive marker for reduced response to chemotherapy.

Knockdown of TRβ enhances resistance to docetaxel and doxorubicin. a, b HCC2185 and HCC202 cells (EV and shTRβ) were treated with DOC, DOX, or CIS for 3 days and IC50 values were calculated using MTT growth assays performed in triplicate. c, d, e HCC2185, HCC202, and MDA-MB-453 cells were treated with DOC 1 or 5 nM (T1 and T5, respectively), DOX10 nM or 100 nM (D10 or D100, respectively) or CIS0.1 or 1 μM (C0.1 or C1, respectively) for 2 months, and endogenous TRβ protein levels were measured using Western blot. GAPDH or RhoGDIα levels were used as loading controls. f, g HCC2185 and HCC202 cells (EV and shTRβ) were treated with DOC 1 nM (T), DOX 100 nM (D), and CIS 1 μM (C) for 4 days, and then Western blots were performed for TRβ, cleaved PARP, PARP, cleaved caspase 3 and caspase 3 expression. GAPDH was used as a loading control
To explore mechanisms underlying resistance induced by TRβ KD, we performed immunoblot analysis to focus on apoptosis induced by chemotherapy. Cysteine-dependent aspartate-directed proteases (caspases) are a family of polypeptides that cleave key cellular proteins to affect the apoptotic cascade [35]. Activated caspase-3 can then cleave poly (ADP-ribose) polymerase (PARP) and block DNA repair [36]. TRβ KD cells dramatically reduced PARP and caspase 3 cleavages with docetaxel and doxorubicin (Fig. 2f, g). Reduction in PARP and caspase-3 cleavage was observed with cisplatin treatment, although no change was seen in IC50 for this drug. These data suggest that low TRβ-expressing tumors may be more resistant to chemotherapy via a reduction in cellular apoptosis.
Cyclic AMP-PKA signaling is involved in TRβ-mediated resistance
We used microarray expression profiling to determine which cellular pathways were altered with TRβ KD in HCC2185 cells. We found 266 probes were differentially expressed between KD and EV (Supplemental Table 1; FDR < 0.1; n = 3 per group). We used DAVID [37] to identify pathways that were enriched in KD cells (148 probes shown in bold). Only the chemokine signaling pathway, including adenylate cyclase 2 which catalyzes the formation of cyclic adenosine monophosphate (cAMP), was significantly enriched (Fig. 3a, p = 0.038, Supplemental Fig. 3 and Table 1). In a pooled analysis of breast cancer patients treated with a number of different chemotherapy regimens, it has been shown that cAMP-mediated signaling, along with several other pathways such as fatty acid and xenobiotic metabolism, were the most consistently associated with resistance to chemotherapy (14), leading us to further examine cAMP signaling in our TRβ KD model.

TRβ-mediated resistance involves cAMP/PKA signaling. a Genes from the top KEGG pathway, chemokine signaling, in TRβ KD cells compared to EV group were identified using DAVID. b and c HCC2185 and HCC202 cells (EV and shTRβ) were treated with DOX, a cAMP inhibitor, alone or in combination for 3 days, and MTT growth assays were performed. *p < 0.05 and **p < 0.01. d and e HCC2185 and HCC202 cells were treated with DOX or the PKA inhibitor H89, alone or in combination and growth assessed using MTT assays. f HCC202 cells were treated with H89 (1 μM) or DOX (D, 200 nM), alone or in combination for 4 days, and analyzed by Western blot analysis. TRβ, pPKA, PKA, cleaved caspase 3, caspase 3, cleaved PARP, and PARP expression was examined using GAPDH as a loading control. g Densitometry of Western blot shown in panel F
Since cAMP is involved in activation of protein kinase A (PKA), we studied their roles in TRβ-mediated resistance. We used a cAMP synthesis inhibitor (Fig. 3b, c), and the competitive PKA inhibitor H89 (Fig. 3d, e), in combination with chemotherapy, in cell growth assays. Both inhibitors were able to significantly decrease growth in combination with doxorubicin in EV HCC2185 and HCC202 cells. The inhibitors were also able to restore sensitivity to doxorubicin in both TRβ KD cells. We did not observe inhibitors affecting docetaxel response (data not shown). As expected TRβ KD increased PKA phosphorylation in both lines (Fig. 3f with the densitometry for PKA, PARP, caspase 3 lanes shown in panel G and Supplemental Fig. 4). H89 in combination with doxorubicin significantly decreased both total and phosphorylated PKA (pPKA), which was most noticeable in the HCC202 TRβ KD cells. Chemotherapeutic drugs inhibit cancer cell growth partly through activation of apoptosis [38]. We therefore performed Western blot analysis to evaluate apoptosis in chemotherapy-treated cells using cleaved caspase-3 and PARP antibodies (Fig. 3f, g). Doxorubicin alone increased cleaved caspase-3, and the cleavage was further increased with doxorubicin in combination with H89 in both HCC202 (Fig. 3f) and HCC2185 cells (Supplemental Fig. 4).These data suggest that inhibition of cAMP/PKA signaling blocks TRβ-mediated resistance. This class of inhibitors might be useful to enhance sensitivity to chemotherapeutics, such as doxorubicin, or perhaps to enable the use of lower, less toxic doses.
TRβ specific agonists enhance sensitivity to chemotherapy through apoptotic mechanisms
T3 is the endogenous TRβ ligand, but it can also bind to TRα and induce serious side effects to both the heart and muscles. Therefore, we tested the effects of two selective TRβ agonists, GC-1 and KB-141, on cell growth [29, 30]. We first determined whether these TRβ agonists could activate canonical thyroid hormone response elements (TRE) in HCC202 EV and KD cells (Supplemental Fig. 5a and b). GC-1 and KB141 both activated TRE activity in HCC202 EV cells, but not KD cells. Steroid receptor ligands can often modulate receptor levels, and we found that both agonists enhanced TRβ protein levels (Fig. 4a and Supplemental Fig. 6a). These data demonstrate that GC-1 and KB-141 may stabilize TRβ receptor protein in these cell lines.

TRβ agonists enhance sensitivity to DOC and DOX. a HCC2185 cells were treated with GC-1, KB-141, or vehicle for 5 days and Western blot analysis performed. GAPDH was used as the loading control. b HCC2185 cells were treated with DOC (0.2 nM) alone, or in combination with GC-1 or KB-141 (0.1, 0.2, 0.4, 0.8, or 1.6 nM) for 9 days, and then growth was assessed using MTT assays. Results are expressed as fold change ± SD relative to vehicle treated cells (**p < 0.01 and ***p < 0.001). c HCC2185 cells were treated with DOX (20, 50, 100, or 200 nM) or GC-1 (1 or 10 nM) or in combination for 6 days, and MTT growth assay was performed to measure cell viability (**p < 0.01 and ***p < 0.001). d HCC2185 cells were treated with DOC (1 or 2 nM) in combination with KB-141 0.1, 1, or 10 nM for 6 days. Western blot analysis was performed for cleaved PARP and total PARP; GAPDH was used as a loading control. e HCC202 cells were treated with DOC, KB141 alone or in combination for 6 days, and analyzed for cleaved PARP, total PARP, cleaved caspase 3, and total caspase 3 levels; GAPDH was used as loading control. f HCC202 cells were treated with DOX or KB-141 alone or in combination. Western blot analysis was performed to measure cleaved PARP, PARP, cleaved caspase 3 and caspase 3. GAPDH was used as loading control
Since higher TRβ levels were associated with chemosensitivity (Fig. 2a, b), and TRβ-specific agonists enhanced TRβ expression (Fig. 4a), we next evaluated the role of GC-1 and KB-141 on docetaxel and doxorubicin sensitivity. GC-1 or KB-141 did not affect basal cell growth, but docetaxel treatment decreased growth by 50 % (Fig. 4b). Docetaxel, in combination with GC-1 or KB-141, significantly decreased cell viability (from 50 to 15 %). GC-1 treatment also decreased proliferation in combination with doxorubicin (Fig. 4c).These data suggest that TRβ-specific agonists enhance chemosensitivity, and therefore lower doses of chemotherapy could be used in patients. These agonists might thus be useful to reduce the known toxicity of chemotherapy.
We next evaluated the effect of TRβ agonists on apoptosis using Western blot analyses. Cells were treated with KB-141, chemotherapy, or in combination. Since KB-141 was the most effective at enhancing TRβ expression and chemosensitivity, we tested it in combination with docetaxel or doxorubicin (Fig. 4d, e, f, respectively). Docetaxel treatment alone enhanced PARP cleavage which was further increased when combined with KB-141. We observed enhanced cleaved caspase 3 when docetaxel was combined with KB-141 (Fig. 4e). KB-141 similarly increased cleaved PARP and cleaved caspase 3 in combination with doxorubicin (Fig. 4f). These cumulative data suggest that enhanced apoptosis underlies the growth inhibitory effects of combined treatment which enhances chemosensitivity.
Discussion
There are few biomarkers which predict chemotherapeutic response in TNBC. NRs are attractive targets because there are agonists and antagonists available, and several TN subtypes overexpress specific NRs such as TRβ. TRβ is a potent nuclear transcription factor which modulates a significant number of genes expressed in brain, liver, and kidney tissues [39]. Herein we hypothesized that TRβ levels could predict sensitivity to chemotherapy. We discovered that low TRβ levels were indeed associated with poor clinical outcomes and enhanced growth of TNBC, and specific KD of TRβ enhanced resistance to both docetaxel and doxorubicin treatment via blockade of apoptosis. In addition, treatment with these agents alone also modulated TRβ levels, thus treatment may initiate this resistance mechanism. Furthermore, we found that TRβ-mediated resistance involved cyclic AMP and PKA signaling, which could be blocked with specific agents. These data suggest that patients with low TRβ expression might be relatively more resistant to specific therapeutic regimens, and might benefit from TRβ-targeted therapies in combination with chemotherapy.
Tumors with high levels of TRβ were associated with elevated levels of other NRs, such as AR and ESR1, but this group does not correspond to the LAR TN subtype defined by Lehmann et al. [20]. High TRβ levels were associated with better outcomes in breast cancer, and we thus hypothesized that elevated levels might influence therapeutic response. Indeed, when we reduced TRβ levels we saw significant effects on chemotherapeutic sensitivity with changes in IC50 values for both docetaxel and doxorubicin. A recent study demonstrated that thyroid hormone levels were lower in chemotherapy-treated breast cancer patients, and in preclinical breast cancer models, treatment with T3 increased drug sensitivity [40]. However, the use of T3 in patients to augment therapeutic response is contraindicated because of toxic effects on the heart. A thyroid hormone analog, eprotirome (KB2115), has been tested in a clinical trial to lower LDL in patients with hypercholesterolemia, and did not demonstrate negative side effects on the heart [41]. We have not tested this agent, but selected two TRβ-specific agonists GC-1 and KB-141 to determine their effects on response. Treatment with these agonists significantly enhanced sensitivity to docetaxel and doxorubicin, and increased cellular. Our data suggest that TRβ-specific agonists in combination with chemotherapy may be useful without initiating deleterious effects of TRα stimulation.
In order to explore downstream mechanisms underlying TRβ’s effects on chemosensitivity, we evaluated gene expression changes after knockdown of TRβ. Chemokine signaling was the only KEGG pathway significantly increased in TRβ KD cells, and cAMP/PKA gene signaling was increased in this pathway by TRβ KD. We confirmed this result demonstrating that TRβ KD cells expressed higher levels of phosphorylated PKA. cAMP activation inhibited doxorubicin-induced apoptosis in leukemia cells through effects on p53 [42]. In ER-negative patients, cAMP-mediated signaling was one of the top pathways associated with chemotherapy response [18]. To test the role of cAMP/PKA activation in our models, we employed a cAMP inhibitor, bupivacaine, and the PKA inhibitor H89 alone and in combination with doxorubicin. Both of these inhibitors restored sensitivity to doxorubicin in all cells. Our data suggest that activation of cAMP/PKA signaling might underlie TRβ’s role in response to chemotherapy. It is tempting to speculate that PKA inhibitors might be useful to enhance chemotherapeutic response in low TRβ-expressing TN tumors.
In summary, low TRβ levels may predict response to specific chemotherapy regimens that are commonly used in TNBC. In addition, TRβ-specific agonists might be used in combination with docetaxel or doxorubicin to enhance sensitivity and to lower doses and decrease toxicity and improve quality of life measures during treatment.
Materials and methods
Reagents and antibodies
Thiazolyl Blue Tetrazolium Bromide (MTT), 3,3′,5 Triiodothyronine and puromycin were obtained from Sigma (St. Louis, MO). MEM, RPMI 1640, DMEM, l-glutamine, penicillin/streptomycin, SeaPlaque™ Agarose was from Lonza (Walkersville, MD). Fetal bovine serum was from Gemini Bio Products, (West Sacramento, CA).Lipofectamine LTX, SuperScript® III Reverse Transcriptase and qPCR probes (ESR1, THRB, COUP-TF1, and GAPDH) were provided by Life Technologies (Grand Island, NY). The renilla luciferase assay kit was from Promega (Madison, WI). TRβ and GAPDH antibodies were from Santa Cruz Biotechnology (Dallas, TX). Total ERα antibody was from Vector Laboratories (Burlingame, CA). AR, c-PARP, PARP, c-Caspase 3, Caspase 3, pPKA, and PKA antibodies were from Cell Signaling Technology (Beverly, MA). ChemiGlow Chemiluminescent Substrate kit was from Protein Simple (Santa Clara, CA). Docetaxel and doxorubicin were from LC Laboratories (Woburn, MA). Cisplatin was from ENZO (Plymouth Meeting, PA). cAMP inhibitor bupivacaine and H89 were obtained from Selleck Chemicals (Houston, TX).
Plasmids
pGIPZ-shTRβ and pGIPZ-EV plasmids were from Thermo Scientific (Pittsburgh, PA). LV113 empty vector and TRβ inducible plasmids were from Genecopoeia (Rockville, MD). TRE renilla reporter was from Switchgear Genomics (Carlsbad, CA). The pLightSwitch-LR plasmid contained synthetic TREs (direct repeat) separated by four nucleotides TGACCCcagctgAGGTCAAGGTCAcaggAGGTCAAGGTCATGACCT. The pLVX-Tet3G plasmid was from Clontech Laboratories, Inc (Mountain View, CA).
Cells and stable transfection
HCC202, HCC38, HCC1569, HCC2185, and ZR75-B were cultured in RPMI 1640 containing 10 % fetal bovine serum. MDA-MB-453 and BT474AZ cells were grown in DMEM containing 10 % fetal bovine serum. MCF-7 cells were cultured in MEM containing 10 % fetal bovine serum. Cell medium contained 0.1 mM nonessential amino acids, and 100 IU/mL penicillin and 100 μg/mL streptomycin. The cells were maintained at 37 °C in the presence of 5 % CO2.
HCC202, HCC2185, and MDA-MB-453 cells were plated in 75 cm2 flasks for one day and then transfected with 10 μg of pGIPZ plasmids containing TRβ shRNA or shEV in regular growth medium. After 48 h transfection, the medium was replaced with 1 μg/ml puromycin for selection. Stable pools were collected for further analysis. The MDA-MB-453 TRβ inducible cell line was created using two steps: parental cells were transduced with pLVX-Tet3G and pools selected with neomycin to stably express Tet protein. pLVX-Tet3G stable pools were transduced with lentivirus vector (LV-113) containing the TRE promoter conjugated with TRβ cDNA and selected with puromycin.
Cell extraction and immunoblot analysis
HCC202, MDA-MB-453, HCC2185, HCC38, HCC1569, and MCF-7 cells were lysed in 100 µl of RIPA buffer containing 50 mM Tris–HCl, 150 mM NaCl, 1 % Nonidet P-40, 0.5 % sodium deoxycholate, 2 mM EDTA, 0.1 % SDS, 1 mM sodium orthovanadate plus 1:100 proteinase inhibitor cocktail III (Calbiochem, La Jolla, CA). Protein concentrations were determined using BCA Protein Assay kits (Pierce, Rockford, IL) according to manufacturer’s directions. Equal amounts of total protein were resolved under denaturing conditions on a 10 % SDS–polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with primary antibodies. For cleaved caspase 3 expression, an 11 % gel was used instead. To ensure equal loading, all membranes were incubated with anti-GADPH or RhoGDIα antibodies. Secondary antibodies were applied to the membranes for 1 h at room temperature and developed with enhanced chemiluminescence reagents (Protein Simple, San Jose, CA).
Anchorage-independent growth assays
Cells (2 × 103/well) were plated in 2 ml of 0.35 % agarose in phenol red-free media, on top of 0.7 % of agarose base layer in 24-well plates. One day after plating, media containing vehicle or treatments as indicated were added to the top layer, and replaced every two days. After 14 days colonies >50 µm in diameter were counted with GelCount. Each condition was performed at least in triplicate.
MTT growth assays
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT, Sigma) assays. Cells (2 × 103 cells/well) were plated in 96-well plates and treated as indicated. 50 µl of MTT stock solution (5 mg/ml in PBS) was added to each well and incubated at 37 °C for 2 h. The media was removed and 100 µl of dimethyl sulphoxide (DMSO) was added to each well. After shaking the plates for 20 min, the absorbance at 570 nM was measured with background subtraction at 655 nM. Each condition of treatment was performed at least in triplicate.
TRE transactivation assays
HCC202 or HCC2185 cells were plated in 24-well plates for one day and transfected with TRE Renilla Reporter and β-Gal. 24 h after transfection, cells were treated for an additional 24 h with T3, GC-1 or KB141 in charcoal-stripped medium. β-galactosidase plasmid was used to normalize the efficiency of the transfection. Renilla luciferase reporter activities were measured by the Renilla Luciferase kit following manufacturer’s protocol. Transfections were performed in triplicate.
Quantitative reverse transcription-PCR (qRT-PCR) assays
Gene expression was evaluated by quantitative reverse transcription-PCR (RT-PCR) method. Cells were grown in 6-well plates to 70–80 % confluency and total RNA extracted using RNeasy kits (Qiagen). 250 ng of total RNA was reverse transcribed in a final volume of 20 µl using Superscript III first-strand kits (Invitrogen), and cDNA was amplified by PCR using the following primers: TRβ, AR, ERα, and GAPDH. Each condition was performed in triplicate.
Microarray analysis
RNA was extracted using RNeasy micro kits (Qiagen). Labeled cRNA was hybridized onto Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix Inc. Santa Clara, CA) in triplicate. Chips were normalized with probeset level estimates of expression previously generated using tools in Bioconductor (www.bioconductor.org) and/or BRB Array tools (http://linus.nci.nih.gov/BRB-ArrayTools.html).
Statistical analyses
Data were analyzed for statistical significance (p < 0.05) using two-tailed Student’s test using GraphPad Prism 5. Standard deviations (SD) are shown. Multivariate Cox proportional hazards model and Kaplan–Meier curve were used for patients DFS analysis in Sabatier et al. dataset [34].
Abbreviations
- AR:
-
Androgen receptor
- CIS or C:
-
Cisplatin
- DOC or T:
-
Docetaxel
- DOX or D:
-
Doxorubicin
- ERα:
-
Estrogen receptor alpha
- EV:
-
Empty vector
- KD:
-
Knockdown
- LAR:
-
Luminal AR
- NRs:
-
Nuclear receptors
- PAM:
-
Prediction analysis of microarrays
- TRβ:
-
Thyroid hormone receptor beta
- TNBC:
-
Triple-negative breast cancer
References
Cuzick J, Sestak I, Bonanni B, Costantino JP, Cummings S, DeCensi A, Dowsett M, Forbes JF, Ford L, LaCroix AZ, Mershon J, Mitlak BH, Powles T, Veronesi U, Vogel V, Wickerham DL, Group SCOBCO (2013) Selective oestrogen receptor modulators in prevention of breast cancer: an updated meta-analysis of individual participant data. Lancet 381(9880):1827–1834. doi:10.1016/S0140-6736(13)60140-3
Early Breast Cancer Trialists’ Collaborative G, Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, Peto R (2011) Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 378(9793):771–784. doi:10.1016/S0140-6736(11)60993-8
Gianni L, Eiermann W, Semiglazov V, Manikhas A, Lluch A, Tjulandin S, Zambetti M, Vazquez F, Byakhow M, Lichinitser M, Climent MA, Ciruelos E, Ojeda B, Mansutti M, Bozhok A, Baronio R, Feyereislova A, Barton C, Valagussa P, Baselga J (2010) Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet 375(9712):377–384. doi:10.1016/S0140-6736(09)61964-4
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948. doi:10.1056/NEJMra1001389
Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, D’Incalci M, Piccolo S, Veronesi A, Zambelli A, Del Sal G, Di Leo A (2013) Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev 39(5):541–550. doi:10.1016/j.ctrv.2012.12.001
Buzdar AU, Singletary SE, Theriault RL, Booser DJ, Valero V, Ibrahim N, Smith TL, Asmar L, Frye D, Manuel N, Kau SW, McNeese M, Strom E, Hunt K, Ames F, Hortobagyi GN (1999) Prospective evaluation of paclitaxel versus combination chemotherapy with fluorouracil, doxorubicin, and cyclophosphamide as neoadjuvant therapy in patients with operable breast cancer. J Clin Oncol 17(11):3412–3417
Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L (2008) Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 26(8):1275–1281. doi:10.1200/JCO.2007.14.4147
von Minckwitz G, Martin M (2012) Neoadjuvant treatments for triple-negative breast cancer (TNBC). Ann Oncol 23(6):vi35–vi39. doi:10.1093/annonc/mds193
Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA (2014) New strategies for triple-negative breast cancer–deciphering the heterogeneity. Clin Cancer Res 20(4):782–790. doi:10.1158/1078-0432.CCR-13-0583
de Hoon JP, Veeck J, Vriens BE, Calon TG, van Engeland M, Tjan-Heijnen VC (1825) Taxane resistance in breast cancer: a closed HER2 circuit? Biochim Biophys Acta 2:197–206. doi:10.1016/j.bbcan.2012.01.001
Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C (2012) Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat Rev 38(7):890–903. doi:10.1016/j.ctrv.2012.02.011
Chan S, Friedrichs K, Noel D, Pinter T, Van Belle S, Vorobiof D, Duarte R, Gil M, Bodrogi I, Murray E, Yelle L, von Minckwitz G, Korec S, Simmonds P, Buzzi F, Gonzalez-Mancha R, Richardson G, Walpole E, Ronzoni M, Murawsky M, Alakl M, Riva A, Crown J, Study G (1999) Prospective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. J Clin Oncol 17(8):2341–2354
Evans TR, Yellowlees A, Foster E, Earl H, Cameron DA, Hutcheon AW, Coleman RE, Perren T, Gallagher CJ, Quigley M, Crown J, Jones AL, Highley M, Leonard RC, Mansi JL (2005) Phase III randomized trial of doxorubicin and docetaxel versus doxorubicin and cyclophosphamide as primary medical therapy in women with breast cancer: an anglo-celtic cooperative oncology group study. J Clin Oncol 23(13):2988–2995. doi:10.1200/JCO.2005.06.156
Nabholtz JM, Senn HJ, Bezwoda WR, Melnychuk D, Deschenes L, Douma J, Vandenberg TA, Rapoport B, Rosso R, Trillet-Lenoir V, Drbal J, Molino A, Nortier JW, Richel DJ, Nagykalnai T, Siedlecki P, Wilking N, Genot JY, Hupperets PS, Pannuti F, Skarlos D, Tomiak EM, Murawsky M, Alakl M, Aapro M et al (1999) Prospective randomized trial of docetaxel versus mitomycin plus vinblastine in patients with metastatic breast cancer progressing despite previous anthracycline-containing chemotherapy. 304 study group. J Clin Oncol 17(5):1413–1424
Jassem J, Pienkowski T, Pluzanska A, Jelic S, Gorbunova V, Mrsic-Krmpotic Z, Berzins J, Nagykalnai T, Wigler N, Renard J, Munier S, Weil C, Central Eastern E, Israel Pacitaxel Breast Cancer Study G (2001) Doxorubicin and paclitaxel versus fluorouracil, doxorubicin, and cyclophosphamide as first-line therapy for women with metastatic breast cancer: final results of a randomized phase III multicenter trial. J Clin Oncol 19(6):1707–1715
Prat A, Lluch A, Albanell J, Barry WT, Fan C, Chacon JI, Parker JS, Calvo L, Plazaola A, Arcusa A, Segui-Palmer MA, Burgues O, Ribelles N, Rodriguez-Lescure A, Guerrero A, Ruiz-Borrego M, Munarriz B, Lopez JA, Adamo B, Cheang MC, Li Y, Hu Z, Gulley ML, Vidal MJ, Pitcher BN, Liu MC, Citron ML, Ellis MJ, Mardis E, Vickery T, Hudis CA, Winer EP, Carey LA, Caballero R, Carrasco E, Martin M, Perou CM, Alba E (2014) Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br J Cancer 111(8):1532–1541. doi:10.1038/bjc.2014.444
Criscitiello C, Azim HA Jr, Schouten PC, Linn SC, Sotiriou C (2012) Understanding the biology of triple-negative breast cancer. Ann Oncol 23(6):vi13–vi18. doi:10.1093/annonc/mds188
Iwamoto T, Bianchini G, Booser D, Qi Y, Coutant C, Shiang CY, Santarpia L, Matsuoka J, Hortobagyi GN, Symmans WF, Holmes FA, O’Shaughnessy J, Hellerstedt B, Pippen J, Andre F, Simon R, Pusztai L (2011) Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J Natl Cancer Inst 103(3):264–272. doi:10.1093/jnci/djq524
Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL (2006) An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 25(28):3994–4008. doi:10.1038/sj.onc.1209415
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767. doi:10.1172/JCI45014
Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell KL, Rugo H, Nabell L, Forero-Torres A, Stearns V, Doane AS, Danso M, Moynahan ME, Momen LF, Gonzalez JM, Akhtar A, Giri D, Patil S, Feigin KN, Hudis CA, Traina TA (2013) Phase II trial of bicalutamide in patients with androgen receptor positive. Hormone receptor negative metastatic breast cancer. Clin Cancer Res. doi:10.1158/1078-0432.CCR-12-3327
Muscat GE, Eriksson NA, Byth K, Loi S, Graham D, Jindal S, Davis MJ, Clyne C, Funder JW, Simpson ER, Ragan MA, Kuczek E, Fuller PJ, Tilley WD, Leedman PJ, Clarke CL (2013) Research resource: nuclear receptors as transcriptome: discriminant and prognostic value in breast cancer. Mol Endocrinol 27(2):350–365. doi:10.1210/me.2012-1265
Martinez-Iglesias O, Garcia-Silva S, Tenbaum SP, Regadera J, Larcher F, Paramio JM, Vennstrom B, Aranda A (2009) Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res 69(2):501–509. doi:10.1158/0008-5472.CAN-08-2198
Guigon CJ, Cheng SY (2009) Novel non-genomic signaling of thyroid hormone receptors in thyroid carcinogenesis. Mol Cell Endocrinol 308(1–2):63–69. doi:10.1016/j.mce.2009.01.007
Brent GA (2012) Mechanisms of thyroid hormone action. J Clin Invest 122(9):3035–3043. doi:10.1172/JCI60047
Cheng SY, Leonard JL, Davis PJ (2010) Molecular aspects of thyroid hormone actions. Endocr Rev 31(2):139–170. doi:10.1210/er.2009-0007
Suhane S, Ramanujan VK (2011) Thyroid hormone differentially modulates Warburg phenotype in breast cancer cells. Biochem Biophys Res Commun 414(1):73–78. doi:10.1016/j.bbrc.2011.09.024
Trost SU, Swanson E, Gloss B, Wang-Iverson DB, Zhang H, Volodarsky T, Grover GJ, Baxter JD, Chiellini G, Scanlan TS, Dillmann WH (2000) The thyroid hormone receptor-beta-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology 141(9):3057–3064
Grover GJ, Mellstrom K, Ye L, Malm J, Li YL, Bladh LG, Sleph PG, Smith MA, George R, Vennstrom B, Mookhtiar K, Horvath R, Speelman J, Egan D, Baxter JD (2003) Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc Natl Acad Sci USA 100(17):10067–10072. doi:10.1073/pnas.1633737100
Johansson L, Rudling M, Scanlan TS, Lundasen T, Webb P, Baxter J, Angelin B, Parini P (2005) Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci USA 102(29):10297–10302. doi:10.1073/pnas.0504379102
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua S, Savage M, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH (2014) Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. doi:10.1158/1078-0432.CCR-14-0432
Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A (2010) Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat Rev 36(3):206–215. doi:10.1016/j.ctrv.2009.12.002
Tibshirani R, Hastie T, Narasimhan B, Chu G (2002) Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA 99(10):6567–6572. doi:10.1073/pnas.082099299
Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, Tallet A, Chabannon C, Extra JM, Jacquemier J, Viens P, Birnbaum D, Bertucci F (2011) A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treat 126(2):407–420. doi:10.1007/s10549-010-0897-9
Chinnaiyan AM (1999) The apoptosome: heart and soul of the cell death machine. Neoplasia 1(1):5–15
Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M (1999) Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 274(33):22932–22940
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57. doi:10.1038/nprot.2008.211
Makin G, Hickman JA (2000) Apoptosis and cancer chemotherapy. Cell Tissue Res 301(1):143–152
Abel ED, Boers ME, Pazos-Moura C, Moura E, Kaulbach H, Zakaria M, Lowell B, Radovick S, Liberman MC, Wondisford F (1999) Divergent roles for thyroid hormone receptor beta isoforms in the endocrine axis and auditory system. J Clin Invest 104(3):291–300. doi:10.1172/JCI6397
Huang J, Jin L, Ji G, Xing L, Xu C, Xiong X, Li H, Wu K, Ren G, Kong L (2013) Implication from thyroid function decreasing during chemotherapy in breast cancer patients: chemosensitization role of triiodothyronine. BMC Cancer 13:334. doi:10.1186/1471-2407-13-334
Ladenson PW, Kristensen JD, Ridgway EC, Olsson AG, Carlsson B, Klein I, Baxter JD, Angelin B (2010) Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med 362(10):906–916. doi:10.1056/NEJMoa0905633
Safa M, Kazemi A, Zand H, Azarkeivan A, Zaker F, Hayat P (2010) Inhibitory role of cAMP on doxorubicin-induced apoptosis in pre-B ALL cells through dephosphorylation of p53 serine residues. Apoptosis 15(2):196–203. doi:10.1007/s10495-009-0417-8
Acknowledgments
The authors would like to thank Dr. Paul Webb from Houston Methodist Research Institute for kindly providing us the TRβ agonists GC-1 and KB-141. We would like to thank Dr. Meng Gao for generating the heat map of Fig. 1a. This work was supported by NIH/NCI R01-CA72038, CPRIT RP120732-P2, and Susan G Komen for the Cure PG12221410.
Conflict of interest
Authors have no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2015_3354_MOESM1_ESM.pptx
Sup. Figure 1.Hierarchical clustering was performed using 41 NR’s to classify227 ER-negative tumors. Contributing receptors were scored using prediction analysis of microarrays [33] across clustered groups; Sup. Figure 2.MDA-MB-453 cells with an inducible TRβ vector were treated with three doses of doxycycline for 48 h,and TRβ expression was evaluated using Western blot. GAPDH was used as positive control. Cells were also treated with doxycycline in combination with DOC or DOX for 6 days and then MTT assays were performed to measure cell viability. Results are expressed as fold change ± SD relative to vehicle control (C) cells.* p < 0.05, **p < 0.01; Sup. Figure 3.DAVID analysis was used to identify significant differential expression of the chemokine signaling pathway. Altered genes included chemokines (CCL2, CX3CL1), Gαi (GNAI1), AC (ADCY2), and PKA (PRKACB) and are denoted with red stars; Sup. Figure 4. HCC2185 cells were treated with H89 or Dox alone or in combination for 4 days and protein expression of TRβ, pPKA, PKA, cleaved caspase 3 and caspase 3 expression evaluated by Western blot analysis; GAPDH was used as a loading control; Sup. Figure 5HCC202 EV or SH cells were transfected with TRE reporterand beta galactosidasefor 24 h, and then treated for an additional 24 h with T3, GC-1, or KB141.Cells were then evaluated for TR transcriptional activity. Results are expressed as fold change ± SD relative to vehicle treated cells. * p < 0.05, **p < 0.01, *** p < 0.001, NS = No Significant; Sup. Figure. 6.A.HCC202 cells were treated with GC-1, KB-141, or vehicle for 5 days and TRβ expression measured using Western blot analysis. GAPDH was used as control. B. HCC202 cells were treated with DOXalone and in combination with GC-1 or KB-141 for 9 days and then MTT assays were formed. Results are expressed as fold change ± SD relative to vehicle treated cells (*** p < 0.001) C. HCC202 cells were treated with DOX or GC-1 alone or in combination for 6 days, and MTT growth assays were performed. * p < 0.05. Supplementary material 1 (PPTX 429 kb)
Rights and permissions
About this article

Cite this article
Gu, G., Gelsomino, L., Covington, K.R. et al. Targeting thyroid hormone receptor beta in triple-negative breast cancer. Breast Cancer Res Treat 150, 535–545 (2015). https://doi.org/10.1007/s10549-015-3354-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3354-y