
Abstract
Aromatase inhibitors (AIs) are used for treatment of estrogen receptor α (ER)-positive breast cancer; however, resistance is a major obstacle for optimal outcome. This preclinical study aimed at identifying potential new treatment targets in AI-resistant breast cancer cells. Parental MCF-7 breast cancer cells and four newly established cell lines, resistant to the AIs exemestane or letrozole, were used for a functional kinase inhibitor screen. A library comprising 195 different compounds was tested for preferential growth inhibition of AI-resistant cell lines. Selected targets were validated by analysis of cell growth, cell cycle phase distribution, protein expression, and subcellular localization. We identified 24 compounds, including several inhibitors of Aurora kinases e.g., JNJ-7706621 and barasertib. Protein expression of Aurora kinase A and B was found upregulated in AI-resistant cells compared with MCF-7, and knockdown studies showed that Aurora kinase A was essential for AI-resistant cell growth. In AI-resistant cell lines, the clinically relevant Aurora kinase inhibitors alisertib and danusertib blocked cell cycle progression at the G2/M phase, interfered with chromosome alignment and spindle pole formation, and resulted in preferential growth inhibition compared with parental MCF-7 cells. Even further growth inhibition was obtained when combining the Aurora kinase inhibitors with the antiestrogen fulvestrant. Our study is the first to demonstrate that Aurora kinase A and B may be treatment targets in AI-resistant cells, and our data suggest that therapy targeting both ER and Aurora kinases may be a potent treatment strategy for overcoming AI resistance in breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Today third-generation aromatase inhibitors (AIs) are recommended as first-line endocrine therapy for postmenopausal women with estrogen receptor α (ER)-positive breast cancer. Steroidal (e.g., exemestane) and non-steroidal (e.g., letrozole and anastrozole) AIs differ in their mechanism of action by binding irreversibly to the substrate site or reversibly to the heme group of the aromatase enzyme, respectively [1]. Despite the efficacy of these compounds, most patients with metastatic disease are either de novo resistant to AIs or will eventually acquire resistance over time. Moreover, in the adjuvant setting, AI resistance also occurs. Thus, AI resistance is one of the most important challenges to overcome in breast cancer treatment, emphasizing the need for development of new treatment strategies.
The molecular mechanisms underlying AI resistance remain largely unknown. So far, preclinical studies using long-term estrogen deprived (LTED) cell models or breast cancer cell lines with ectopic overexpression of the aromatase enzyme have suggested that ER signaling along with growth factor signaling, e.g., through EGFR, HER2, IGFR, Src, and PI3K/Akt/mTOR, is involved in AI-resistant cell growth [2–6], and differences in resistance mechanisms to steroidal and non-steroidal AIs have been reported [5]. Clinical studies have shown that combination of AI treatment with kinase inhibitors may enhance clinical benefit, e.g., the BOLERO study, where combination of exemestane and the mTOR inhibitor everolimus demonstrated increased efficacy compared to exemestane alone with respect to progression-free survival [7].
Several clinical trials are currently evaluating the potential of Aurora inhibitors in cancer treatment [8, 9]. The Aurora kinases (Aurora A/B/C) constitute a family of highly conserved serine/threonine kinases, which play important roles in cell cycle control as they regulate progression through mitosis and cytokinesis [10, 11]. Aurora A and B have both been linked to cancer [8, 9]. While Aurora A coordinates centrosome maturation, assembly of the bipolar spindle and chromosome separation, Aurora B regulates chromosome condensation, the spindle checkpoint, and cytokinesis [11]. We have recently shown that Aurora A [12] and Aurora B (Larsen et al., unpublished data) are involved in resistance to antiestrogen therapy, and expression of the kinases are associated with poor survival in breast cancer patients who have received adjuvant tamoxifen treatment.
In this study, we present data from a functional kinase inhibitor screen using different letrozole- and exemestane-resistant cell lines, established by long-term AI treatment of MCF-7 breast cancer cells grown under conditions at which cell growth was dependent on conversion of testosterone to estradiol via the endogenous aromatase enzyme [13, 14]. We identified Aurora A and B as potential new treatment targets for AI-resistant breast cancer cells.
Materials and methods
Cell lines and culture conditions
The MCF-7 cell line was originally obtained from the Human Cell Culture Bank (Mason Research Institute, Rockville, MD, USA). A subline, MCF-7/S0.5 (MCF-7), adapted to grow with low amount of fetal calf serum (FCS) [15, 16] was used as parental cells for the AI-resistant cell lines MCF-7/S0.5/LetR-1 (LetR-1), MCF-7/S0.5/LetR-3 (LetR-3), MCF-7/S0.5/ExeR-1 (ExeR-1), and MCF-7/S0.5/ExeR-3 (ExeR-3), which were established by selection of surviving colonies from long-term AI treatment (10−6 M letrozole or 10−7 M exemestane (Selleck Chemicals)) of the MCF-7/S0.5 cells grown in DMEM/F12 medium supplemented with 10 % newborn calf serum (NCS) (Life Technologies, Carlsbad, CA, USA), 10−7 M testosterone, 6 ng/mL insulin, and 2.0 mM GlutaMAX (Sigma-Aldrich, St. Louis, MO, USA) [14]. The AI-resistant cell lines were maintained in DMEM/F12 medium with 10 % NCS, 10−7 M testosterone, 6 ng/mL insulin, 2.0 mM GlutaMAX, and their respective AI [14].
Kinase inhibitor screen
The kinase inhibitor screen (L1200 from Selleck Chemicals) was performed as previously described [12]. Cells were seeded in 96-well plates in medium containing 10 % NCS and 10−7 M testosterone (MCF-7), and for AI-resistant cell lines, containing their respective AI. Cells were left for 2 days before treatment for 5 days with 1 µM inhibitor. DMSO (0.1 %)-treated controls were included in each plate. Cell viability was assayed using CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI, USA) and measured using Varioscan Flash platereader (Thermo Scientific, Waltham, MA, USA).
Cell growth assays
Cells were seeded in standard growth medium as described above. For combination studies, AI-resistant cell lines were withdrawn from their respective AI 1 week before onset of the experiment. Cells were allowed to adhere for 2 days before treatment for 5 days with the indicated concentrations of JNJ-7706621, barasertib, alisertib, danusertib (Selleck Chemicals), and fulvestrant (ICI 182,780; Tocris Bioscience, Bristol, UK). Cell number was determined by crystal violet staining as described previously [17]. All experiments were repeated at least twice with similar results.
Western blot analyses
Cells were seeded in 6-well plates and treated with inhibitors as indicated. Harvesting of cells and Western blot analysis was performed as previously described [18]. Antibodies targeting the following proteins were used; Aurora A (4718) from Cell Signaling Technology (Danvers, MA, USA), Aurora B (AJ1069a) from Abgent (San Diego, CA, USA), and Hsp70 (MS-482-PO) from Thermo Scientific. Western blots were performed at least twice with similar results. Quantitation was done using Image J.
Knockdown of Aurora kinases
Cells were transfected with 20 nM of each siRNA using Amaxa Cell Line Nucleofector Kit V and Nucleofector (Lonza, Basel, Switzerland) according to the manufacturer’s instructions. Mission siRNA duplexes (Sigma-Aldrich) were used for knockdown (siAuroraA#1, SASI_Hs01_00079240; siAuroraA#2; SASI_Hs01_00079241; siAuroraB#1, SASI_Hs01_00076963; siAuroraB#2, esiRNA EHU001471). Scramble sequence control siRNA was Mission Universal Negative Control siRNA (SIC001, Sigma-Aldrich). After transfection, cells were seeded for Western blot analysis or cell growth assays.
Flow cytometry
Cell cycle analysis was done as described previously [19]. Cells were fixed in 2 % formaldehyde followed by permeabilization in ethanol, blocking in 0.5 % BSA/PBS, and incubation with AlexaFluor488-conjugated phospho-Histone-H3Ser10 antibody (3465, Cell Signaling Technology). Cells were incubated with 20 µg/ml propidium iodide and 40 µg/ml RNase A and analyzed using a BD FACSVerse flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA) and FlowJo Software v.X (Tree Star Inc., Ashland, OH, USA).
Immunofluorescence microscopy
Cells were seeded in their standard growth medium and treated with kinase inhibitors as indicated. Cells were fixed with methanol for 15 min at −20 °C and washed with PBS/1 % FBS before blocking for 1 h with PBS/10 % FBS/0.5 % Triton-X-100. Incubation was done overnight at 4 °C with antibodies against Aurora A (4718) or Aurora B (AJ1069a). Secondary antibody was Alexa-Fluor-488 goat-anti-rabbit antibody (Molecular Probes, Life Technologies). Slides were incubated with 1 μg/ml Hoechst 33342 (Sigma-Aldrich) and mounted using Fluorescent Mounting Medium (DAKO, Glostrup, Denmark). Pictures were captured using an Olympus IX71 (Tokyo, Japan). For evaluation of Aurora A and B positive cells, pictures were blinded and double-scored. Positive cells were defined as cells with intensive nuclear green staining, and % positive cells were calculated relative to the total number of nuclei, evaluated by Hoechst staining, from the same sample.
Statistical analyses
Group comparisons were done using a two-tailed t test with Bonferroni adjusted P values for multiple testing. P < 0.05 was considered statistically significant.
Results
A kinase inhibitor screen identifies compounds exerting preferential growth inhibition of AI-resistant cell lines
We have recently established a new cell culture model for acquired resistance to AIs from the ER-positive breast cancer cell line MCF-7, grown under conditions where cell growth was dependent on conversion of androgen to estrogen via the endogenous aromatase enzyme [13, 14]. To identify putative new treatment targets in AI-resistant breast cancer, we performed a kinase inhibitor screen using the parental MCF-7 cells, two letrozole-resistant cell lines, LetR-1 and LetR-3 (Fig. 1a), and two exemestane-resistant cell lines, ExeR-1 and ExeR-3 (Fig. 1b). The screen included 195 different kinase inhibitors and treatment of cells with 1 µM inhibitor for five days resulted in identification of 24 hits (Online Resource 1), defined as compounds with at least 2-fold statistically significant (P < 0.05) growth inhibition of one or more AI-resistant cell line compared with MCF-7. Table 1 presents selected hits, which exerted at least 20 % higher growth inhibition of AI-resistant cell lines than MCF-7. Notably, several of the selected hits target Aurora kinases, which we have recently shown to be important for growth of tamoxifen-resistant breast cancer cells [12]. Moreover, other common targets of the hits include cyclin-dependent kinases (CDKs), Akt, vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and Abl (Table 1 and Online Resource 1).

Identification of compounds preferentially inhibiting growth of AI-resistant breast cancer cell lines. The parental cell line MCF-7 together with four different AI-resistant cell lines (LetR-1, LetR-3, ExeR-1 and ExeR-3) were seeded in 96-well plates and treated in triplicate with a kinase inhibitor library, comprising 195 different compounds. After 5 days of treatment with 1 μM kinase inhibitor, cell number was assessed using CellTiter-Glo luminescent cell viability assay. Volcano plots were generated by plotting the growth inhibitory effects in a the letrozole-resistant cell lines and b the exemestene-resistant cell lines relative to MCF-7 cells (x axis) against the P values from t test comparison of the effect on the AI-resistant cell lines versus MCF-7 (y axis). Hits were identified using a two-fold cutoff value for the relative growth inhibition and a P value <0.05 (located between the blue lines). The inhibitors JNJ-7706621 and barasertib are shown in the figure
The Aurora inhibitors JNJ-7706621 and barasertib inhibit growth of AI-resistant cell lines and cause accumulation of cells in the G2/M cell cycle phase
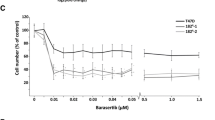
Because the Aurora A/B and CDK inhibitor JNJ-7706621 exerted pronounced growth inhibition of all four AI-resistant cell lines, and five of the other selected inhibitors targeted Aurora kinases, we decided to use the broad inhibitor JNJ-7706621 and the selective Aurora B inhibitor barasertib for further studies. Dose–response growth assays were performed with the two compounds in MCF-7, LetR-1, LetR-3, ExeR-1, and ExeR-3 cells (Fig. 2a, b). Treatment with 0.5 µM JNJ-7706621 resulted in significantly more growth inhibition of ExeR-1 cells compared with MCF-7, while 1 µM and 1.5 µM JNJ-7706621 preferentially inhibited growth of all four AI-resistant cell lines compared with MCF-7 (P < 0.05) (Fig. 2a). Treatment with 0.05–1 µM barasertib resulted in statistically significant preferential growth inhibition of ExeR-1 cells compared with MCF-7 (Fig. 2b). For ExeR-3, LetR-1, and LetR-3 cell lines, preferential growth inhibition was found using 0.05–0.1 µM barasertib. Aurora A and B are specifically expressed during the G2/M phase [11], and we have previously observed increased levels of Aurora A and B and accumulation of cells in G2/M phase upon treatment of tamoxifen-resistant MCF-7 cells with JNJ-7706621 [12]. Similar to these results, treatment of MCF-7, LetR-1, and ExeR-1 cells with 1 µM JNJ-7706621 caused increased levels of Aurora A and B (Fig. 2c). Quantitation of protein level after 24 h treatment with JNJ-7706621 revealed 7.3, 2.5, and 1.4 fold increase of Aurora A expression and 3.1, 2.2, and 12.5 fold increase of Aurora B expression in MCF-7, LetR-1, and ExeR-1 cells, respectively. Accumulation of cells in the G2/M cell cycle phase was also observed (Fig. 2d). Notably, MCF-7 cells were not as affected by JNJ-7706621 treatment as the AI-resistant cell lines, where the most pronounced accumulation of cells in G2/M was found in ExeR-1 cells after treatment with JNJ-7706621 for 48 h (Fig. 2d). Barasertib treatment for up to 24 h did not cause increase in Aurora A and B levels; instead a tendency of decreased Aurora B protein level was observed in AI-resistant cell lines upon treatment with barasertib for 24 h (Fig. 2c.). This is in line with previous data showing that barasertib results in destabilization of Aurora B protein [20]. Cell cycle analysis revealed that barasertib induced accumulation of cells in G2/M, where the most severe accumulation was found for AI-resistant cell lines (Fig. 2e).

Effects of the Aurora/CDK inhibitor JNJ-7706621 and the Aurora B inhibitor barasertib on cell growth, expression of Aurora A and B and cell cycle phase distribution. MCF-7 and AI-resistant cell lines were treated for 5 days with the indicated concentrations of a JNJ-7706621 or b barasertib. The cell number was measured by a colorimetric assay and expressed as percent of DMSO-treated control cells. Error bars indicate SD. At least two independent experiments were performed with reproducible results and a representative experiment is shown. Stars indicate significant difference from MCF-7 cells (P < 0.05). c For Western blot analyses, MCF-7 cell line and AI-resistant cell lines were grown in their standard medium followed by treatment with 0.1 % DMSO (control), 1 µM JNJ-7706621, or 1 µM barasertib for 1 or 24 h. Hsp70 was used as a loading control. FACS analysis of the cell cycle phase distribution of MCF-7, LetR-1, and ExeR-1 cells was performed after treatment with d 1 μM JNJ-7706621 and e 0.05 µM barasertib
In the above-mentioned experiments, the respective AI was present in the growth medium when the AI-resistant cell lines were treated with the kinase inhibitors. Omission of AI from the resistant cells was performed for 1 week and then JNJ-7706621, and alisertib treatment was applied in the presence and absence of increasing concentrations of AI. The Aurora inhibitors had little effect on growth of MCF-7 cells, whereas growth of MCF-7 cells decreased with increasing concentration of AIs (Online Resource 3). Pronounced growth inhibition of LetR-1 and ExeR-1 was seen with Aurora inhibitors without AI, and addition of increasing concentration of AIs had no further growth inhibitory effect. Thus, Aurora inhibitors do not resensitize the AI-resistant cells to AI treatment.
Aurora kinases are upregulated in AI-resistant cell lines and important for cell growth
Although the expression levels of Aurora kinases varied between experiments, presumably due to their regulation during the cell cycle, we generally observed that both Aurora A and B were expressed at higher levels in the AI-resistant cells lines compared with MCF-7 (Fig. 3a). To investigate the role of the Aurora kinases for growth of AI-resistant cell lines, we depleted MCF-7, LetR-1, and ExeR-1 cells of either Aurora A or B using different siRNA constructs. Two siRNAs targeting Aurora A (siAuroraA#1 and siAuroraA#2) both resulted in efficient knockdown of Aurora A protein (Fig. 3b). Aurora A knockdown caused severe growth inhibition of AI-resistant cells, while only a minor reduction in growth was observed for MCF-7 (Fig. 3c), suggesting that Aurora A is essential for growth of the AI-resistant cells. Two different Aurora B siRNA constructs (siAuroraB#1 and siAuroraB#2) were used; however, none of the constructs resulted in complete knockdown of Aurora B protein (Fig. 3b). The partial Aurora B knockdown obtained using the siAuroraB#1 construct resulted in minor growth inhibition of AI-resistant cell lines, and for ExeR-1 cells, the difference from the effect on MCF-7 cells was statistically significant (Fig. 3d).

Knockdown of Aurora A and B by siRNA in MCF-7 and AI-resistant cell lines. a Protein expression of Aurora A and B in MCF-7, LetR-1, LetR-3, ExeR-1, and ExeR-3, grown in their standard medium, was measured by Western blot analysis. b MCF-7, LetR-1, and ExeR-1 cells were transfected with control siRNA (siControl) or independent siRNAs targeting Aurora A (siAurA#1 and #2) or Aurora B (siAurB#1 and #2). Protein expression of Aurora A and B was measured by Western blot analysis. Hsp70 was used as a loading control. c and d Cell growth assays were performed 96 h after transfection with the siRNA constructs. The cell number was measured by a colorimetric assay and expressed as percent of siControl-transfected cells. Error bars indicate SD. Stars denote statistically significant difference from MCF-7 cells (P < 0.05)
The clinically relevant Aurora inhibitors alisertib and danusertib preferentially inhibit growth of AI-resistant cell lines
Based on our results showing that Aurora A is important for growth of AI-resistant cell lines (Fig. 3), we decided to explore the effect of alisertib (MLN8237), a highly potent and selective Aurora A inhibitor currently undergoing phase II/III trials [21], on AI-resistant cell lines. Furthermore, we included the clinically relevant (phase II) compound danusertib (PHA-739358), a potent inhibitor of Aurora A, and also a modest inhibitor of Abl, RET, TrkA, FGFR1, and Aurora B/C [22]. Dose–response growth assays in MCF-7, LetR-1, and ExeR-1 cells showed that alisertib (0.05–0.5 µM) and danusertib (0.1–0.5 µM) preferentially inhibited growth of the AI-resistant cell lines compared with MCF-7 (Fig. 4a, b). Western blot analysis showed increased level of Aurora A in MCF-7 (11.7 fold), LetR-1 (3.6 fold), and ExeR-1 (1.7 fold) after 24 h treatment with alisertib, indicative of accumulation of cells in G2/M (Fig. 4c). Alisertib had no significant effect on Aurora B expression in the three cell lines. For danusertib, no change in protein expression of Aurora A and B was observed in MCF-7 or the resistant cell lines. Cell cycle analysis showed that both alisertib (Fig. 4d) and danusertib (Fig. 4e) treatment resulted in preferential accumulation of LetR-1 and ExeR-1 cells in the G2/M cell cycle phase upon treatment for 24 and 48 h.

Effects of the Aurora A inhibitor alisertib and the Aurora A/B/C inhibitor danusertib on cell growth, expression of Aurora A and B and cell cycle phase distributions. MCF-7 and AI-resistant cell lines were treated for 5 days with indicated concentrations of a alisertib or b danusertib. The cell number was measured by a colorimetric assay and expressed as percent of DMSO-treated control cells. Error bars indicate SD. Three independent experiments were performed with reproducible results and a representative experiment is shown. Stars indicate significant difference from MCF-7 cells (P < 0.05). c For Western blot analyses, MCF-7, LetR-1, and ExeR-1 were grown in their standard medium followed by treatment with 0.1 % DMSO (control), 0.1 µM alisertib, or 0.1 µM danusertib for 1 or 24 h. Hsp70 was used as a loading control. Cell cycle analysis of MCF-7, LetR-1 and ExeR-1 cells was performed after treatment with d 0.05 µM alisertib or e 0.05 µM danusertib for 24 and 48 h
Alisertib and danusertib block cell cycle progression at mitosis and prevent correct localization of Aurora kinases during metaphase
To explore the mechanisms by which the Aurora inhibitors exerted their action, we analyzed the mitosis-specific Aurora B target phospho-Histone-H3Ser10 by flow cytometry. After treatment for 24 h, JNJ-7706621 and barasertib treatment both completely abrogated Histone-H3-phosphorylation, indicative of inhibition of Aurora B activity and blocked transition from G2 to M phase (Fig. 5). In contrast, alisertib and danusertib treatment resulted in pronounced accumulation of cells in M phase, in particular for AI-resistant cell lines, indicative of cell cycle arrest at mitosis (Fig. 5). We evaluated Aurora A and B localization upon treatment for 24 h with the different inhibitors. In dividing control cells, Aurora A was localized to the two spindle poles during metaphase, while Aurora B was associated with the aligned chromosomes (Fig. 6a), as previously shown [23]. Upon treatment with JNJ-7706621 or barasertib, metaphase cells were infrequently observed, and in JNJ-7706621-treated cells, Aurora A and B localization was comparable to control cells, while barasertib caused perturbed Aurora B localization and misaligned chromosomes, especially in the resistant cell lines (Fig. 6a). Upon treatment with alisertib, no cells with aligned chromosomes could be observed; however, we noticed an increase in cells with condensed DNA and strong dispersed staining for Aurora A, while Aurora B staining was associated with the condensed DNA. Upon treatment with danusertib, aligned chromosomes were observed (Fig. 6a). We quantified cells with strong Aurora A and B staining and found that the percentage of positive cells were significantly increased in the two resistant cell lines compared with MCF-7 upon treatment with alisertib or danusertib (Fig. 6b, c). Notably, the fraction of Aurora kinase positive cells in DMSO-treated controls was significantly higher for AI-resistant cell lines than for MCF-7, supporting the observed increased expression of Aurora A and B (Fig. 3a).

Effects of JNJ-7706621, barasertib, alisertib, or danusertib treatment on cell cycle phase distribution. MCF-7, LetR-1 and ExeR-1 were treated for 24 h with 0.1 % DMSO, 1 µM JNJ-7706621, 0.05 µM barasertib, 0.05 µM alisertib, or 0.05 µM danusertib. Cells were harvested, fixed, and stained with AlexaFlour488-conjugated phospho-Histone-H3Ser10-antibody and propidium iodide and analyzed by flow cytometry. The upper right quadrants show phospho-histone-H3-positive cells with 4 N DNA, representing M phase cells. The lower right quadrants represent G2 phase cells, whereas the lower left quadrants show cells in G1/S phase. Percentages of cells in each phase were quantified using FlowJo Software and indicated in the figure. The experiments were performed twice with similar results

Localization of Aurora A and B after treatment with JNJ-7706621, barasertib, alisertib, or danusertib. MCF-7, LetR-1, and ExeR-1 were treated for 24 h with 0.1 % DMSO, 1 µM JNJ-7706621, 0.05 µM barasertib, 0.05 µM alisertib, or 0.05 µM danusertib. The cells were fixed with methanol and stained with antibodies against Aurora A and B. Nuclei were stained with Hoechst and fluorescent imaging was used to identify Aurora A and Aurora B positive cells. a Pictures were captured using an Olympus IX71. For evaluation of b Aurora A and c Aurora B positive cells, pictures were blinded and double-scored and percentage of positive cells were calculated relative to the total number of nuclei, evaluated by Hoechst staining, from the same sample
Combination of an Aurora inhibitor with fulvestrant is superior to treatment with either of the compounds alone
Both MCF-7 cells and the AI-resistant cell lines responded to treatment with the ER down modulator fulvestrant (0.1 µM) (Fig. 7a–c), indicative of ER-dependent cell growth. To determine whether the Aurora inhibitors could be combined with ER-targeted therapy, cells were treated with 1 µM JNJ-7706621 (Fig. 7a), 0.05 µM alisertib (Fig. 7b), or 0.05 µM danusertib (Fig. 7c) alone or in combination with 0.1 µM fulvestrant. For AI-resistant cell lines, the combination treatment resulted in significantly more growth inhibition compared with fulvestrant or Aurora inhibitor treatment alone. When combining alisertib with fulvestrant, significantly more growth inhibition compared with either of the compounds alone was also observed for MCF-7 cells (Fig. 7b); however, the growth inhibitory effect of combined treatment was more severe for AI-resistant cell lines compared with MCF-7. Together, these data suggest a beneficial effect of combined treatment targeting both ER and Aurora kinases in AI-resistant breast cancer cells.

Effects of JNJ-7706621, alisertib, or danusertib in combination with fulvestrant on cell growth in MCF-7 and AI-resistant cell lines. LetR-1 and ExeR-1 were withdrawn from their respective AI and were, together with MCF-7, treated for 5 days with a 1 µM JNJ-7706621, 0.1 µM fulvestrant and combination, b 0.05 µM alisertib, 0.1 µM fulvestrant and combination, or c 0.05 µM danusertib, 0.1 µM fulvestrant and combination. The cell number was measured by a colorimetric assay and expressed as percent of DMSO-treated control cells. Error bars indicate SD. Two independent experiments were performed with reproducible results, and a representative experiment is shown. Stars indicate significant difference both when comparing the combined treatment with fulvestrant and the combined treatment with the Aurora inhibitor alone (P < 0.05)
Discussion
A major obstacle in current breast cancer treatment is development of resistance against AIs. Clinical trials showing benefit from combining kinase inhibitors with AIs suggest that therapy against activated signaling pathways may be a way forward to prevent or postpone resistance [7]. Preclinical models are important tools to better understand the molecular pathways driving AI resistance. In this study, we used a cell culture model in which testosterone by conversion to estradiol via the endogenous aromatase enzyme could stimulate growth of MCF-7 cells [13]. At 1 nM testosterone concentration, which corresponds to physiological concentration in postmenopausal women [24], we found a 4-fold growth stimulation. This supports the clinical relevance of the MCF-7 model used to establish the resistant cell lines by clonal selection of cells, surviving long-term treatment with either letrozole or exemestane [14]. By application of a large functional kinase inhibitor screen, we identified novel treatment targets in the AI-resistant cells, e.g., Aurora A and B, CDK1/2/9, CHK1, Akt, PDGFR, Src, Abl, and DUB. We focused our analyses on the Aurora kinases, as they were targets in all four investigated AI-resistant cell lines, and several Aurora inhibitors exerted pronounced and preferential inhibition of growth of AI-resistant cell lines compared to MCF-7 cells. Furthermore, Aurora A has recently been discovered as a target in tamoxifen-resistant breast cancer cells [12, 25].
Our data suggest that in particular Aurora A plays a pivotal role for growth of AI-resistant cells. Significant growth inhibition was observed with the Aurora A and B inhibitor JNJ-7706621, with the clinically relevant selective Aurora A inhibitor alisertib and with the potent Aurora A inhibitor danusertib when grown in medium with AI. Omission of AI did not affect this growth inhibition, indicating that the inhibitors do not resensitize AI-resistant cells to AIs. The severe growth inhibition of AI-resistant cell lines after knockdown with specific siRNAs against Aurora A also confirms the important role of Aurora A for AI-resistant cell growth. We demonstrated increased expression of Aurora A in AI-resistant cell lines compared with MCF-7. We have recently demonstrated that Aurora A plays a role for growth of tamoxifen-resistant breast cancer cells, which utilize ER for growth, presumably through ligand-independent ER activation [12, 26]. It has been shown that Aurora A can phosphorylate ER, thus contributing to its ligand-independent activation [25]. In the LTED cell model, it was demonstrated that ligand-independent activation of ER can drive AI-resistant cell growth [27]. Our AI-resistant model responds to fulvestrant treatment, indicating that the resistant cells require ER for growth. It is therefore possible that activated protein kinases, such as Aurora A, cause ligand-independent activation of ER, thereby circumventing the need for estrogen synthesis by the aromatase enzyme upon acquisition of resistance. However, since we observed additional growth inhibition from combining Aurora inhibitors with fulvestrant, we presume that the inhibitors also target ER-independent mechanisms. Notably, the combination growth assays with Aurora inhibitor and fulvestrant were performed without AIs; however, similar results were observed in the presence of AIs (data not shown). These results show that AI is not required for the beneficial effect of combined treatment, suggesting that patients progressing on AI therapy may benefit from switching to fulvestrant treatment in combination with an Aurora inhibitor. Clinical trials are warranted to disclose this and at present, patients with ER-positive metastatic breast cancer are being recruited for a clinical trial combining fulvestrant and alisertib (NCT02219789).
Our results indicate that Aurora B may also play a role in AI resistance. Barasertib, JNJ-7706621, and danusertib all inhibited growth of the resistant cell lines. Although we could not obtain complete knockdown of Aurora B, we observed a small but significant growth inhibition of the ExeR-1 cell line, suggesting involvement of Aurora B in AI-resistant cell growth. Furthermore, the Aurora B target phospho-Histone-H3Ser10 was completely abrogated upon treatment with barasertib and JNJ-7706621, validating that the compounds inhibit the kinase activity of Aurora B. We observed an increase in polyploid cells (>4 N) and severe mitotic defects upon barasertib treatment for up to 96 h (Online Resource 2), indicative of endoreduplication and impaired mitotic checkpoint resulting from Aurora B inhibition [10]. These defects were more pronounced for AI-resistant cell lines than MCF-7, suggesting that AI-resistant cells are more dependent on Aurora B for correct cell division. The clinical relevance of our results is supported by our previous studies, showing that Aurora A [12] and Aurora B (Larsen et al., unpublished data) are both potential biomarkers for tamoxifen resistance, and evaluation of Aurora A expression in tumor material from patients who have received adjuvant letrozole treatment is presently ongoing. Moreover, barasertib, alisertib, and danusertib are currently in clinical trials as new cancer therapeutics, which so far show promising results [9].
Based on our preclinical studies, we conclude that Aurora A, Aurora B, and ER play important roles for AI-resistant cell growth and may be targets for treatment. The observed overexpression of Aurora A and B in AI-resistant cell lines indicates that Aurora kinases may have potential as biomarkers, which in combination with ER can be used to select patients for the combined treatment.
References
Chumsri S, Howes T, Bao T, Sabnis G, Brodie A (2011) Aromatase, aromatase inhibitors, and breast cancer. J Steroid Biochem Mol Biol 125(1–2):13–22
Ghosh D, Griswold J, Erman M, Pangborn W (2009) Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 457(7226):219–223
Chan CM, Martin LA, Johnston SR, Ali S, Dowsett M (2002) Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. J Steroid Biochem Mol Biol 81(4–5):333–341
Kazi AA, Gilani RA, Schech AJ, Chumsri S, Sabnis G, Shah P, Goloubeva O, Kronsberg S, Brodie AH (2014) Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res 16(1):R15
Masri S, Phung S, Wang X, Wu X, Yuan YC, Wagman L, Chen S (2008) Genome-wide analysis of aromatase inhibitor-resistant, tamoxifen-resistant, and long-term estrogen-deprived cells reveals a role for estrogen receptor. Cancer Res 68(12):4910–4918
Liu S, Meng X, Chen H, Liu W, Miller T, Murph M, Lu Y, Zhang F, Gagea M, Arteaga CL, Mills GB, Meric-Bernstam F, Gonzalez-Angulo AM (2014) Targeting tyrosine-kinases and estrogen receptor abrogates resistance to endocrine therapy in breast cancer. Oncotarget 5(19):9049–9064
Beaver JA, Park BH (2012) The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol 8(6):651–657
Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W (2010) Aurora kinase inhibitors–rising stars in cancer therapeutics? Mol Cancer Ther 9(2):268–278
Goldenson B, Crispino JD (2014) The aurora kinases in cell cycle and leukemia. Oncogene. doi:10.1038/onc.2014.14
Nair JS, Ho AL, Tse AN, Coward J, Cheema H, Ambrosini G, Keen N, Schwartz GK (2009) Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol Biol Cell 20(8):2218–2228
Vader G, Lens SMA (2008) The Aurora kinase family in cell division and cancer. Biochim Biophys Acta 1786(1):60–72
Thrane S, Pedersen AM, Thomsen MBH, Kirkegaard T, Rasmussen BB, Duun-Henriksen AK, Lænkholm AV, Bak M, Lykkesfeldt AE, Yde CW (2014) A kinase inhibitor screen identifies Mcl-1 and Aurora kinase A as novel treatment targets in anti-estrogen-resistant breast cancer cells. Oncogene. doi:10.1038/onc.2014.351
Sonne-Hansen K, Lykkesfeldt AE (2005) Endogenous aromatization of testosterone results in growth stimulation of the human MCF-7 breast cancer cell line. J Steroid Biochem Mol Biol 93(1):25–34
Hole S, Pedersen AM, Hansen SK, Lundqvist J, Yde CW, Lykkesfeldt AE (2015) New cell culture model for aromatase inhibitor-resistant breast cancer shows sensitivity to fulvestrant treatment and cross-resistance between letrozole and exemestane. Int J Oncol. doi:10.3892/ijo.2015.2850
Briand P, Lykkesfeldt AE (1984) Effect of estrogen and antiestrogen on the human breast cancer cell-line MCF-7 adapted to growth at low serum concentration. Cancer Res 44(3):1114–1119
Lykkesfeldt AE, Madsen MW, Briand P (1994) Altered expression of estrogen-regulated genes in a tamoxifen-resistant and ICI 164,384 and ICI 182,780 sensitive human breast cancer cell line, MCF-7/TAMR-1. Cancer Res 54(6):1587–1597
Lundholt BK, Briand P, Lykkesfeldt AE (2001) Growth inhibition and growth stimulation by estradiol of estrogen receptor transfected human breast epithelial cell lines involve different pathways. Breast Cancer Res Treat 67(3):199–214
Pedersen AM, Thrane S, Lykkesfeldt AE, Yde CW (2014) Sorafenib and nilotinib resensitize tamoxifen resistant breast cancer cells to tamoxifen treatment via estrogen receptor alpha. Int J Oncol 45(5):2167–2175
Yde CW, Olsen BB, Meek D, Watanabe N, Guerra B (2008) The regulatory beta-subunit of protein kinase CK2 regulates cell-cycle progression at the onset of mitosis. Oncogene 27(37):4986–4997
Gully CP, Zhang F, Chen J, Yeung JA, Velazquez-Torres G, Wang E, Yeung SC, Lee MH (2010) Antineoplastic effects of an Aurora B kinase inhibitor in breast cancer. Mol Cancer 9:42
Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, Leroy PJ, Gershman RE, Silva MD, Germanos MS, Bolen JB, Claiborne CF, Sells TB (2011) Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 17(24):7614–7624
Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, Arrigoni C, Soncini C, Alli C, Patton V, Marsiglio A, Ballinari D, Pesenti E, Fancelli D, Moll J (2007) PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther 6(12 Pt 1):3158–3168
Scrittori L, Skoufias DA, Hans F, Gerson V, Sassone-Corsi P, Dimitrov S, Margolis RL (2005) A small C-terminal sequence of Aurora B is responsible for localization and function. Mol Biol Cell 16(1):292–305
Lonning PE, Eikesdal HP (2013) Aromatase inhibition 2013: clinical state of the art and questions that remain to be solved. Endocr Relat Cancer 20(4):R183–R201
Zheng XQ, Guo JP, Yang H, Kanai M, He LL, Li YY, Koomen JM, Minton S, Gao M, Ren XB, Coppola D, Cheng JQ (2013) Aurora-A is a determinant of tamoxifen sensitivity through phosphorylation of ERalpha in breast cancer. Oncogene. doi:10.1038/onc.2013.444
Thrane S, Lykkesfeldt AE, Larsen MS, Sorensen BS, Yde CW (2013) Estrogen receptor alpha is the major driving factor for growth in tamoxifen-resistant breast cancer and supported by HER/ERK signaling. Breast Cancer Res Treat 139(1):71–80
Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, Jiang A, Smith RA, Maira SM, Manning HC, Gonzalez-Angulo AM, Mills GB, Higham C, Chanthaphaychith S, Kuba MG, Miller WR, Shyr Y, Arteaga CL (2011) ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov 1(4):338–351
Acknowledgments
We would like to thank Birgit Reiter for excellent technical assistance and Dr. Monika Marine Mortensen for help with FACS analysis. This work was supported by Grants from the Danish Cancer Society, Danish Cancer Research Foundation, Astrid Thaysen’s Foundation, Wedell-Wedellsborg’s Foundation, Harboe Foundation, and A Race Against Breast Cancer.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Stine Hole and Astrid M. Pedersen have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material. Online Resource 1. Data for hits identified in the kinase inhibitor screen. Triplicate samples of parental MCF-7 cells and AI-resistant cells (LetR-1, LetR-3, ExeR-1 and ExeR-3) were treated with 1.0 µM of the indicated kinase inhibitors for five days. The effect of each compound was determined by a cell viability assay. Hits were defined as compounds exerting at least two-fold statistically significant growth inhibition of one or more AI-resistant cell line compared with MCF-7 cells (indicated in red for the individual cell lines). Inhibitory effect (± SD) and P-values are shown.Online Resource 2. Cell division defects induced by barasertib. a) MCF-7, LetR-1, and ExeR-1 were treated for 48 days with 0.1 % DMSO or 0.05 µM barasertib, and cell cycle analyses were done by flow cytometry. Gating of single cells was done, and the DNA content was measured as the intensity of the propidium iodide signal. Percentage of cells with > 4 N DNA content was quantified using FlowJo Software and indicated in the figure. b ) Upon treatment of cells for 96 h with 0.1 % DMSO or 0.05 µM barasertib, 1 μg/ml Hoechst 33342 was added to the medium and fluorescence pictures of live cells were captured using an Olympus IX71 microscope. Scale bar; 10 µm.Online Resource 3. Dose – response growth experiments with MCF - 7 and AI - resistant cell lines with AI in the presence and absence of JNJ - 7706621 or alisertib. Aromatase inhibitor was withdrawn from the medium of the resistant cell lines one week before start of the experiment. MCF-7, LetR-1, and ExeR-1 cells were treated with increasing concentrations of letrozole or exemestane as indicated with or without a) 0.5 μM JNJ-7706621 or b) 0.05 µM alisertib. Cell number was determined after five days by a colorimetric assay and expressed as percentage of the control cells (0.1 % EtOH treated). Error bars represent SD of the mean of at least three replicate values. Asterisks (*) indicate significant growth inhibition compared with cells grown without aromatase inhibitor (p < 0.05).
Rights and permissions
About this article

Cite this article
Hole, S., Pedersen, A.M., Lykkesfeldt, A.E. et al. Aurora kinase A and B as new treatment targets in aromatase inhibitor-resistant breast cancer cells. Breast Cancer Res Treat 149, 715–726 (2015). https://doi.org/10.1007/s10549-015-3284-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-015-3284-8