
Abstract
Interfering oncogenic STAT3 signaling is a promising anti-cancer strategy. We examined the efficacy and drug mechanism of an obatoclax analog SC-2001, a novel STAT3 inhibitor, in human breast cancer cells. Human breast cancer cell lines were used for in vitro studies. Apoptosis was examined by both flow cytometry and western blot. Signaling pathways were assessed by western blot. In vivo efficacy of SC-2001 was tested in xenograft nude mice. SC-2001 inhibited cell growth and induced apoptosis in association with downregulation of p-STAT3 (Tyr 705) in breast cancer cells. STAT3-regulated proteins, including Mcl-1, survivin, and cyclin D1, were repressed by SC-2001. Over-expression of STAT3 in MDA-MB-468 cells protected cells from SC-2001-induced apoptosis. Moreover, SC-2001 enhanced the expression of protein tyrosine phosphatase SHP-1, a negative regulator of STAT3. Furthermore, the enhanced SHP-1 expression, in conjunction with increased SHP-1 phosphatase activity, was mediated by upregulated transcription by RFX-1. Chromatin immunoprecipitation assay revealed that SC-2001 increased the binding capacity of RFX-1 to the SHP-1 promoter. Knockdown of either RFX-1 or SHP-1 reduced SC-2001-induced apoptosis, whereas ectopic expression of RFX-1 increased SHP-1 expression and enhanced the apoptotic effect of SC-2001. Importantly, SC-2001 suppressed tumor growth in association with enhanced RFX-1 and SHP-1 expression and p-STAT3 downregulation in MDA-MB-468 xenograft tumors. SC-2001 induced apoptosis in breast cancer cells, an effect that was mediated by RFX-1 upregulated SHP-1 expression and SHP-1-dependent STAT3 inactivation. Our study indicates targeting STAT3 signaling pathway may be a useful approach for the development of targeted agents for anti-breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is a major issue in women’s health worldwide [1, 2]. Breast cancers that are characterized by lack of expression of the estrogen receptor, progesterone receptor, or human epidermal growth factor receptor type 2 (HER2) have been classified as triple-negative breast cancer (TNBC) [2]. TNBC accounts for 10–20 % of invasive breast cancers and is characterized by occurrence in younger women, aggressive clinical behaviors with a high recurrence rate, high incidence of metastasis, and poor prognosis despite chemosensitivity at the initial stage [2, 3]. Currently, there are no well-established specific therapeutic targets for TNBC. Increasing knowledge of the biology of TNBC has led to promising studies using therapies such as EGFR-targeted agents, anti-angiogenics, and PARP inhibitors [4]. Despite promising data from preclinical studies, there are still no validated targeted therapies available for TNBC patients. Therefore, there is a pressing need to uncover the molecular mechanisms of TNBC. Revealing new compounds that target these mechanisms may advance the development of TNBC treatments.
Management of breast cancer has been greatly influenced by the availability of novel-targeted therapies as a result of increasing biological knowledge and the identification of new molecular targets. Apoptosis-resistance is one of the major reasons for breast cancer progression and tumorigenesis [5]. The anti-apoptotic proteins of the B cell lymphoma 2 (Bcl-2) family have been shown to be overexpressed and dysregulated in breast cancer [6], and have emerged as attractive targets for novel anti-cancer drugs. Obatoclax is a BH3 mimetic that can interact with anti-apoptotic Bcl-2 family proteins at their BH3-binding grooves and disrupt the interaction of the anti-apoptotic proteins and bind pro-apoptotic proteins [7]. In preclinical studies of breast cancer cells, obatoclax has shown a synergistic effect when used with lapatinib, a targeted therapy for HER2-positive breast cancer, to induce apoptotic and autophagic cell death in human breast cancer cells [8–10].
Several oncogenic transcription factors, such as signal transducer and activator of transcription 3 (STAT3), can regulate the transcription of the anti-apoptotic Bcl-2 family proteins. STAT3 has emerged as a potential anti-cancer target as it is crucial in the regulation of genes involved in cell proliferation and survival, and is constitutively activated in common human cancers, including breast cancer [11, 12]. In cancer cells, constitutively activated STAT3 directly contributes to tumorigenesis, invasion, and metastasis [12]. STAT3 has been shown to play an important role in breast carcinogenesis and is often overexpressed in primary breast tumors [13]. STAT3 can be phosphorylated by tyrosine kinase which is activated by cytokines, growth factors, or hormones. Upon tyrosine phosphorylation, STAT3 homodimerizes or heterodimerizes with STAT1, then translocates to the nucleus, and activates transcription of target genes by binding to the consensus sequence of the promoters [12]. STAT3 regulates expression of numerous apoptosis-related proteins, including Bcl-2, Bcl-xL, Mcl-1, survivin, and cyclin D1 [14]. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice [15]. Scientists have shown that reduction of STAT3 activity has synergistic effect with doxorubicin in breast cancers [16]. Notably, STAT3 signaling is downregulated by protein tyrosine phosphatases, such as the SH2 domain-containing tyrosine phosphatase family (SHP-1 and SHP-2), and protein tyrosine phosphatase 1B (PTP-1B). These phosphatases reduce STAT3 activation directly by dephosphorylation of STAT3 [17]. Therefore, activity of protein tyrosine phosphatases may be critical for the regulation of STAT3 activity in cancer cells. A previous study revealed that SHP-1 expression was confined to a well-defined subset of high-grade breast tumors, including HER2-positive or estrogen receptor-negative tumors [18]. Recently, Esposito et al. reported that SHP-1 expression correlates with imatinib sensitivity in chronic myeloid leukemia cells [19]. Moreover, several investigational agents with anti-cancer potential have been shown to increase either SHP-1 expression or its activity and suppress p-STAT3 signaling [20–26]. Our previous results also suggest that SHP-1-dependent STAT3 inhibition is a major target of sorafenib and its analogs in hepatocellular carcinoma and breast cancer cells [27, 28]. Collectively, these data suggest that inhibiting STAT3 by enhanced SHP-1 phosphatase function is a potential therapeutic target in breast cancers.
Previously, we reported the efficacy of SC-2001, a novel obatoclax derivative, in hepatocellular carcinoma cells [29]. SC-2001 is structurally related to the Mcl-1 inhibitor obatoclax [30]. We showed that SC-2001 not only inhibits the protein–protein interactions between Mcl-1 and Bak but also down-regulates Mcl-1 by reducing its transcription [29]. We further discovered that SC-2001 can down-regulate p-STAT3 and showed better antitumor effects than obatoclax in hepatocellular carcinoma cells [29]. Here, we examined the efficacy and drug mechanism of SC-2001 in breast cancer cells including TNBC cells.
Materials and methods
Reagents and antibodies
Synthesis, purification, and characterization of SC-2001
SC-2001 ((Z)-2-((3-methoxy-2H-pyrrol-2-ylidene)methyl)-1H-pyrrole) compounds were synthesized and purified in the medicinal chemistry laboratory at the Institute of Biopharmaceutical Sciences, National Yang-Ming University, Taiwan. The detailed process has been described in our previous study [29]. The structures of SC-2001 and obatoclax are shown in Supplementary Fig. 1. SC-2001 compounds were subjected to nuclear magnetic resonance and mass spectrometry for structure and molecular weight characterization. The molecular weight of SC-2001 calculated by high-resolution mass spectrometry for C18H14BrN3O (M+H+) was 368.0393.
Other reagents and antibodies
Sodium vanadate and specific SHP-1 inhibitor were purchased from Cayman Chemical (Ann Arbor, MI, USA). Antibodies for immunoblotting such as cyclin D1 and PARP were purchased from Santa Cruz Biotechnology (San Diego, CA, USA). Other antibodies such as survivin, phospho-STAT3 (Tyr705), STAT3, and SHP-1 were from Cell Signaling (Danvers, MA, USA).
Cell culture
The HCC-1937, MDA-MB-231, MDA-MB-468, MDA-MB-453, and MCF-7 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). All breast cancer cells were maintained in DMEM medium supplemented with 10 % fetal bovine serum, 0.1 mM nonessential amino acids, 2 mM l-glutamine, 100 units/mL penicillin G, 100 µg/mL streptomycin sulfate, and 25 µg/mL amphotericin B in a humidified incubator in an atmosphere of 5 % CO2 at 37 °C in air. Lysates of breast cancer cells were treated with drugs at the indicated concentrations for various periods of time.
Cell viability and proliferation of breast cancer cell lines in vitro
Cell viability and proliferation of breast cancer cells treated with or without SC-2001 were assessed by colorimetric assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Cells were plated in a 96-well plate in 100 μL DMEM per well and cultured for up to 24 h. Cells were incubated for 4 h at 37 °C with MTT; after incubation, the medium was removed and the cells were treated with DMSO for 5 min. Viability was evaluated by ultraviolet absorption spectrum at 550 nm with a Microplate Reader Model 550 (Bio-Rad, Richmond, CA, USA).
Apoptosis analysis
Drug-induced apoptotic cell death was assessed by the following three methods: (a) measurement of apoptotic cells by flow cytometry (sub-G1 analysis), (b) Western blot analysis of PARP cleavage, and (c) detection of cytoplasmic histone-associated DNA fragmentation with a Cell Death Detection ELISA kit (Roche Diagnostics Mannheim, Germany). ELISA was conducted according to the manufacturer’s instructions. Briefly, breast cancer cells were treated with DMSO or SC-2001 at the indicated dose for 24 h, and the specific enrichment of oligonucleosomes released into the cytoplasm was quantified by cell death ELISA.
Dual luciferase assay
After transfection with firefly luciferase reporter construct and reference pCMV-renilla luciferase plasmid for 48 h, cells were collected and lysed with passive lysis buffer. The lysate was placed into glass tube, and promoter activity was analyzed by dual luciferase assay according to the instruction manual.
Gene knockdown using siRNA
Smart-pool siRNA, including control (D-001810-10), SHP-1, and RFX-1, were all purchased from Dharmacon (Chicago, IL, USA). Briefly, cells were transfected with siRNA (final concentration, 100 nM) in 6-well plates using the lipid-mediated transfection with Lipofectamine2000 (Invitrogen, Life technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. After 48 h, the medium was replaced and the breast cancer cells were incubated with SC-2001, harvested, and separated for western blot analysis and apoptosis analysis by flow cytometry as described previously [28].
MDA-MB-468 cells with ectopic expression of STAT3
STAT3 cDNA was purchased from Origene (Rockville, MD, USA), and the vector backbone is pCMV6-Entry (C-terminal Myc and DDK Tagged). MDA-MB-468 cells with ectopic expression of STAT3 derived from a single-stable clone were prepared for in vitro assay for STAT3 target validation. Briefly, following transfection, cells were incubated in the presence of Geneticin (G418, 0.78 mg/mL, Invitrogen). After 8 weeks of selection, surviving colonies, i.e., those arising from stably transfected cells, were selected and individually amplified.
Phosphatase activity assays
The RediPlate 96 EnzChek Tyrosine Phosphatase Assay Kit (R-22067) was used for SHP-1 activity assay (Molecular Probes, Carlsbad, CA, USA). Briefly, breast cancer cell protein extracts were incubated with anti-SHP-1 antibody in immunoprecipitation buffer overnight. Protein G-Sepharose 4 Fast Flow (GE Healthcare, Piscataway, NJ, USA) was added to each sample followed by incubation for 3 h at 4 °C with rotation and then assayed for phosphatase activity.
Colony-forming assays
Colony-forming assays were prepared in triplicate wells with breast cancer cells. Cells were seeded in 6-well plates (~1,000–5,000 cells per well) and subjected to the indicated treatments, with the drug being removed to terminate the treatment. Two weeks later, plates were washed in PBS, fixed with 100 % methanol, and stained with a filtered solution of crystal violet (5 % w/v). After washing with tap water, the colonies were counted both manually (by eye) and digitally using a ColCount TM plate reader (Oxford Optronics, Oxford, England).
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed according to the protocol of the EZ ChIP chromatin immunoprecipitation and EZ-Zyme Chromatin Prep Kit (Upstate Biotechnology, Lake Placid, NY, USA) according to the manufacturer’s description. Briefly, after cross-linking with 17.5 % paraformaldehyde, cells were washed with phosphate-buffered saline and lysed in lysis buffer. The DNA was fragmented to about 200–500 base pairs by the EZ-Zyme™. Approximately, 5 × 106 cells were used per ChIP assay and the resulting DNA fragments were incubated with 2 μg RFX1 antibodies (NBP1-52652) (Novus Biologicals, Littleton, CO, USA), which were generated from rabbit, or non-specific rabbit IgG (Millipore). The immunoprecipitated products were washed sequentially with low-salt immune complex wash buffer, high-salt immune complex wash buffer, LiCl immune complex wash buffer, and twice with TE buffer. The chromatin was eluted from the agarose by incubating with elution buffer (1 % SDS, 100 mM NaHCO3); the DNA–protein complexes were reversely cross-linked by high-salt solution containing 200 mM NaCl at 65 °C for at least 5 h. To eliminate contaminations of proteins and RNAs, the mixture was treated with 10 mg RNase A at 37 °C for 30 min and then treated with protease K for 2 h at 45 °C. Finally, the precipitated DNA was recovered using the spin column provided in the ChIP kit, and eluted with 50 ml elution buffer. PCR reaction was conducted using Taq DNA polymerase (MyTaq). Two microliters of the precipitated DNA was used as the template. The sequences of the primers used in the ChIP assay were as follows: 5′-CCTCTTGCAGGTGTCCTTAAG-3′, and 5′-TGGAAAGGCAGAGGGAATCAG-3′.
Xenograft tumor growth
Female NCr athymic nude mice (4–6 weeks of age) were obtained from the National Laboratory Animal Center (Taipei, Taiwan, ROC). The mice were housed in groups and maintained in an SPF-environment. All experimental procedures using these mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital. Each mouse was inoculated subcutaneously in the dorsal flank with 2 × 106 breast cancer cells suspended in 0.1 mL serum-free medium containing 50 % Matrigel (BD Biosciences, Bedford, MA, USA) under isoflurane anesthesia. Tumors were measured using calipers and their volumes calculated using a standard formula: width2 × length × 0.52. When tumors reached 100–200 mm3, mice received SC-2001 (20 mg/kg/every other day), or vehicle alone (control group). On termination of treatment, mice were sacrificed and xenografted tumors were harvested and assayed for molecular events by western blot analysis and for SHP-1 activity.
Immunohistochemical staining
Paraffin-embedded breast cancer tissue sections (4 μm) on poly-1-lysine-coated slides were deparaffinized and rinsed with 10 mM Tris–HCl (pH 7.4) and 150 mM sodium chloride. Paroxidase was quenched with methanol and 3 % hydrogen peroxide. Slides were then placed in 10 mM citrate buffer (pH 6.0) at 100 °C for 20 min in a pressurized heating chamber. After incubation with 1:50 dilution of rabbit monoclonal antibody to SHP-1 (ab32559) (Abcam, Cambridge, MA, USA), or with 1:50 dilution of rabbit polyclonal antibody to RFX-1 (NBP1-52652) (Novus Biologicals, Littleton, CO, USA), or with 1:50 dilution of rabbit polyclonal antibody to p-STAT3 (Tyr705) (ab30646) (Abcam, Cambridge, MA, USA) for 1 h at room temperature, slides were thoroughly washed three times with phosphate-buffered saline. Bound antibodies were detected using the EnVision Detection Systems Peroxidase/DAB, Rabbit/Mouse kit (Dako, Glostrup, Denmark). The slides were then counterstained with hematoxylin.
This study was approved by the ethics committee of the Institutional Review Board of Taipei Veterans General Hospital. Informed consent was obtained from all sample donors at time of their donation in accordance with the Declaration of Helsinki.
Statistical analysis
Data are expressed as mean ± SD or SE. Statistical comparisons were based on nonparametric tests and statistical significance was defined at p < 0.05. All statistical analyses were performed using SPSS for Windows software, version 12.0 (SPSS, Chicago, IL, USA).
Results
SC-2001 induces apoptosis in breast cancer cells including triple-negative breast cancer cells
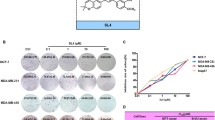
To investigate the antitumor effect of SC-2001 on breast cancer cells, we first assessed growth inhibition in response to SC-2001 treatment in a panel of five human breast cancer cell lines: triple-negative breast cancer (TNBC) cells HCC-1937, MDA-MB-231, and MDA-MB-468; human epidermal growth factor receptor type 2 (HER2)-overexpressing cells MDA-MB-453; and estrogen receptor-positive cells MCF-7. Cell viability was determined by MTT assay after treatment for 48 h at the indicated doses. As shown in Fig. 1a, SC-2001 significantly reduced cell viability in a dose-dependent manner in all tested cell lines. We next assessed the apoptotic effect of SC-2001 in human breast cancer cell lines. The results showed that SC-2001 induced apoptotic effects in a dose- and time-dependent manner (Fig. 1b, c). Of note, SC-2001 elicited obvious cell death in a relatively low dose range (less than 2 μM). These data indicate that human breast cancer cell lines, including TNBC cells, are sensitive to the cytotoxic activity of SC-2001.

SC-2001 induces apoptosis in triple-negative breast cancer cells. a Dose-escalation effects of SC-2001 on cell viability in breast cancer cell lines. Cells were exposed to SC-2001 at the indicated doses for 48 h, and cell viability was assessed by MTT assay. Points, mean (n = 3); bars, SD. b Upper panel, dose-escalation effects of SC-2001 on apoptosis in breast cancer cell lines. Cells were exposed to SC-2001 at the indicated doses for 48 h. Lower panel, time-dependent effects of SC-2001 on apoptosis in breast cancer cell lines. Cells were exposed to SC-2001 at the indicated doses for 24, 48, and 72 h. Apoptotic cells were analyzed by flow cytometry. Columns, mean (n = 3); bars, SD. c Dose-escalation effects of SC-2001 on apoptosis in triple-negative breast cancer cell lines MDA-MB-468, MDA-MB-231, and HCC-1937. Cells were exposed to SC-2001 at the indicated doses for 24 h. Apoptotic cells were analyzed by detection of cytoplasmic histone-associated DNA fragmentation with the Cell Death Detection ELISA kit (Roche Diagnostics Mannheim, Germany). Columns, mean (n = 3); bars, SD
Downregulation of p-STAT3 contributes to the apoptotic effects of SC-2001 in TNBC cells
Drug-induced apoptosis was evidenced by the activation of PARP cleavage (Fig. 2a). Our previous report showed that inhibition of STAT-3 and downregulation of Mcl-1 are key factors in SC-2001-induced apoptosis [29]. To elucidate the mechanism by which SC-2001 inhibits cell growth and induces apoptosis in TNBC cell lines, we examined the effects of SC-2001 on STAT-3 and its downstream death signaling molecules Mcl-1, cyclin D1, and survivin. SC-2001 downregulated p-STAT3 (Y705) as well as Mcl-1, cyclin D1, and survivin in a dose-dependent manner in the three tested TNBC cell lines without altering of total protein levels of STAT3 (Fig. 2a). We then examined whether SC-2001 affected the transcription activity of STAT3 in TNBC cells. Our data showed that SC-2001 decreased the DNA-binding activity of STAT3 (Fig. 2b). Taken together, these data demonstrate that SC-2001 inactivates STAT3, reduces its DNA binding activity, and subsequently suppresses transcription of the downstream molecules Mcl-1, cyclin D1, and survivin in TNBC cells. To demonstrate the role of STAT3 in SC-2001-induced apoptosis in TNBC cells, STAT3 was ectopically transfected into MDA-MB-468 cells. Overexpression of STAT3 significantly reduced the effects of SC-2001 on apoptosis in MDA-MB-468 cells (Fig. 2c). These results confirm the importance of STAT3 inhibition in mediating the effect of SC-2001 in TNBC cells.

Downregulation of p-STAT3 contributes apoptotic effects of SC-2001 in triple-negative breast cancer cells. a dose-escalating effects of SC-2001 on p-STAT3 and STAT3-regulated proteins MCl-1, survivin, cyclin D1, and cleavage PARP-1. Cells were exposed to SC-2001 at indicated doses for 24 h. Cell lysates were prepared and assayed for these molecules by western blotting. Western blot data are representative of three independent experiments. Apoptosis is evidenced by cleavage of PARP-1. b Effects of SC-2001 on STAT3 transcription activity. STAT3-binding region was cloned into Luc reporter and was transfected into MDA-MB-231 and MDA-MB-468 cells. Cells were then exposed to SC-2001 at 2 μM for 6 h. The firefly luciferase activity was evaluated and normalized by Renilla luciferase. Columns, mean; bars, SD (n = 3). *p < 0.05. c Protective effect of STAT3 on apoptosis induced by SC-2001 in MDA-MB-468 cells. Cells (wild-type or ectopic expression of STAT3) were exposed to SC-2001 at 2 μM for 48 h. Apoptotic cells were analyzed by flow cytometry. Columns, mean; bars, SD (n = 3). *p < 0.05
SHP-1 mediates the effects of SC-2001 on p-STAT3
Protein phosphatase SHP-1 is a tyrosine phosphatase that is known to mediate the phosphorylation of JAK/STAT signaling [17]. To further delineate the crucial regulator of SC-2001-induced apoptosis and dephosphorylation of STAT3 in TNBC cells, we analyzed the protein expression of SHP-1 in TNBC cells after SC-2001 treatment. Our data revealed that SC-2001 upregulated the protein level of SHP-1 in a time-dependent manner in all three tested TNBC cell lines (Fig. 3a). SC-2001 increased the transcription of SHP-1 in TNBC cells in a time-dependent manner (Fig. 3b). Moreover, SC-2001 increased SHP-1 activity significantly in the tested cells (Fig. 3c). To examine whether increased SHP-1 activity accounted for the effects of SC-2001 induction in TNBC cells, we first assessed the effects of sodium vanadate, a general protein tyrosine phosphatase inhibitor and a specific SHP-1 inhibitor PTP inhibitor III on SC-2001-induced downregulation of p-STAT3 and apoptosis. Both these two inhibitors affect the activity of SHP-1. Sodium vanadate reversed the effects of SC-2001 on p-STAT3 and DNA fragmentation (Fig. 3d, 1st). Similarly, we found that the SHP-1-specific inhibitor also reversed SC-2001-induced inhibition of p-STAT3 and cell death (Fig. 3d, 2nd). Moreover, knock-down of SHP-1 by using siRNA significantly abolished the effects of SC-2001 on p-STAT3 and DNA fragmentation as well as the colony formation ability (Fig. 3d, 3rd and 4th). Furthermore, either sodium vanadate or specific SHP-1 inhibitor alone did not affect cell viability; despite these two agents efficiently inhibit SHP-1 activity (Supplementary Fig. 2). Taken together, these data indicate that SC-2001 induces p-STAT3 downregulation and mediates apoptosis through a SHP-1-dependent mechanism in TNBC cells.

SHP-1 mediates effects of SC-2001 on p-STAT3. a Time-dependent effects of SC-2001 on SHP-1 expression in association with p-STAT3 expression in MDA-MB-231, MDA-MB-468, and HCC-1937 cells. Cells were exposed to SC-2001 at 2 μM for 6, 9, and 12 h. Cell lysates were prepared and assayed for these molecules by western blotting. Western blot data are representative of three independent experiments. b Time-dependent effects of SC-2001 on SHP-1 mRNA levels in MDA-MB-231, MDA-MB-468, and HCC-1937 cells. Cells were exposed to SC-2001 at 2 μM for 6, 9, 12, and 18 h. SHP-1 mRNA extracts were assayed by quantitative RT-PCR. Columns, mean; bars, SD (n = 3). *p < 0.05, **p < 0.01. c Time-dependent effects of SC-2001 on SHP-1 activity in MDA-MB-231, MDA-MB-468, and HCC-1937 cells. Cells were treated with SC-2001 at 2 μM for 6 and 9 h, and then cell lysates were analyzed by phosphatase activity assay. Columns, mean; bars, SD (n = 6). *p < 0.05, **p < 0.01. D, SHP-1-dependent inhibition of STAT3 mediates the antitumor effects of SC-2001. 1st, and 2nd, inhibition of SHP-1 reverses effects of SC-2001 on apoptosis. 1st, sodium vanadate, a non-specific phosphatase inhibitor. 2nd, PTP inhibitor III, a specific SHP-1 inhibitor. MDA-MB-468 cells were pretreated with sodium vanadate (25 μM) or specific SHP-1 inhibitor (50 μM) for 120 min and then treated with SC-2001 at 2 μM for 24 h. Apoptotic cells were assayed by Cell Death Detection ELISA kit. Columns, mean; bars, SD (n = 3). *p < 0.05. 3rd, silencing SHP-1 by siRNA reduces the effects of SC-2001 on p-STAT3 inhibition and apoptosis in MDA-MB-468 cells. Cells were transfected with control siRNA (scrambled) or SHP-1 siRNA for 48 h then treated with SC-2001 at 2 μM for another 24 h. Apoptotic cells were assayed by Cell Death Detection ELISA kit. Columns, mean; bars, SD (n = 3). *p < 0.05. 4th, silencing SHP-1 by siRNA reduces the inhibitory effects of SC-2001 on colony-forming ability of MDA-MB-468 cells. Cells were treated with SC-2001 at 2 μM for 24 h and then were prepared for colony-forming assays as described in “Materials and methods.” Colonies were scored after 14 days of culture. Columns, mean; bars, SD (n = 6). *p < 0.05
SC-2001 leads to cell death by modulating RFX-1/SHP-1 signaling pathway
A previous report showed that RFX-1 is a major transcription factor of SHP-1 in breast cancer cells [31]. To investigate whether RFX-1 is involved in the regulation of SC-2001-induced SHP-1 expression, first, the expression level of RFX-1 in SC-2001-treated cells was examined. RFX-1 and SHP-1 were found to be upregulated in a dose-dependent manner in SC-2001-treated MDA-MB-468 cells (Fig. 4a). To demonstrate the involvement of RFX-1 in SHP-1 transcription in SC-2001-treated cells, chromatin IP of RFX-1 was performed. As shown in Fig. 4b, the binding capacity of RFX-1 to the SHP-1 promoter was obviously increased in the SC-2001-treated cells. Furthermore, silencing of RFX-1 by RNAi reduced the effects of SC-2001 on SHP-1 expression, apoptosis, and colony formation (Fig. 4c, e, left). Ectopic expression of RFX-1 in MDA-MB-468 cells increased SHP-1 expression; treating RFX-1-overexpressing cells with SC-2001 further increased SHP-1 expression and induced more apoptosis than that of any single treatment (Fig. 4d). Concurrently treating RFX-1-overexpressing cells with SC-2001 almost completely abolished colony formation ability (Fig. 4e, right). Finally, we treated three TNBC cell lines with SC-2001 in an earlier time-dependent manner and analyzed the mRNA levels of RFX-1 as well as the cell viability (Supplementary Fig. 3). As shown in Supplementary Fig. 3, the mRNA expressions of RFX-1 in three TNBC cell lines are already upregulated, whereas the cell viability is not obviously affected, indicating the RFX-1/SHP-1 pathway is the cause of SC-2001-mediated cell death. Taken together, these data suggest that RFX-1 regulates SHP-1 transcription and plays an important role in mediating SC-2001-induced cell death in TNBC cells.

SC-2001 leads to cell death by modulating RFX-1/SHP-1 signaling pathway. a Dose-dependent effects of SC-2001 on SHP-1 and RFX-1 expression. MDA-MB-468 cells were treated with SC-2001 in a dose-dependent manner. Cell lysates were prepared and assayed for these molecules by western blotting. Western blot data are representative of three independent experiments. b MDA-MB-468 cells were treated with 2 μM of SC-2001 for 12 h. DNA from control and SC-2001-treated MDA-MB-468 cells was immunoprecipitated with RFX-1 or rabbit IgG antibody and captured by protein A-agarose beads. The RFX-1-binding site fragment in the SHP-1 promoter was detected by PCR in ChIP samples. C, RFX-1 was involved in the SC-2001-induced cell death effect. MDA-MB-468 cells were transfected with control siRNA or RFX-1 siRNA for 48 h then treated with SC-2001 at 2 μM for 24 h. Apoptotic cells were analyzed by Cell Death Detection ELISA kit (Roche Diagnostics Mannheim, Germany). d RFX-1 overexpression can enhance the effect of SC-2001. MDA-MB-468 cells were transfected with vector control or RFX-1 overexpression plasmid for 48 h and then treated with SC-2001 at 2 μM for 24 h. Apoptotic cells were analyzed by Cell Death Detection ELISA kit (Roche Diagnostics Mannheim, Germany). e Left, knockdown of RFX-1 can reduce the inhibitory effect of SC-2001 on colony-forming ability of cells. Cells were transfected with control si-RNA and RFX-1 si-RNA and then treated with SC-2001 at 2 μM for 24 h. Then the cells were prepared for colony formation assay. Right, overexpression of RFX-1 enhanced the inhibitory effect of SC-2001 on colony-forming ability of cells. Cells were transfected with vector control or RFX-1 overexpression plasmid for 48 h and then treated with SC-2001 at 2 μM for 24 h. Then the cells were prepared for colony formation assay. For c–e, Columns, mean; bars, SD (n = 3). *p < 0.05
Therapeutic evaluation of SC-2001 on TNBC xenograft tumor growth in vivo
To verify the drug effect in vivo, we assessed the effects of SC-2001 on MDA-MB-468 xenograft tumors. Tumor-bearing mice were treated with vehicle or SC-2001 orally at a dose of 20 mg/kg every other day for three weeks. As a result, mice receiving SC-2001 showed remarkable tumor growth inhibition without exhibiting any notable body weight loss (Fig. 5a, b). To verify the mechanism identified in vitro, western blots of p-STAT3, SHP-1, and RFX-1 were examined in isolated tumors at termination of treatment. Consistent with the in vitro findings, SC-2001 downregulated p-STAT3 without affecting STAT3 expression, and increased the protein levels of RFX-1 and SHP-1 in MDA-MB-468 tumors (Fig. 5c). In addition, p-STAT3 activity was shown to be significantly lower in the SC-2001-treated tumors than the control group (Fig. 5d). Moreover, treatment with SC-2001 induced SHP-1 activity in tumors (Fig. 5e). Taken together, these data indicate that SC-2001 demonstrates anti-TNBC tumor growth activity in vivo by means of SHP-1-mediated STAT3 inactivation.

Therapeutic evaluation of SC-2001 on MDA-MB-468 xenograft tumor growth in vivo. a Growth curves of MDA-MB-468 tumors treated with vehicle, and SC-2001 (20 mg/kg/every other day). Female NCr athymic nude mice (4–6 weeks of age) were treated with SC-2001 (20 mg/kg body weight) per os every other day. Controls received vehicle. Points, mean (n = 6); bars, SD. b Body weight of xenograft mice bearing MDA-MB-468 tumors during the in vivo experiment. Points, mean (n = 6); bars, SD. c Western blot analysis of p-STAT3, STAT3, SHP-1, and RFX-1 in MDA-MB-468 xenograft tumors. Tumors were harvested at 21 days after treatment and assayed for molecular events by western blotting and also for p-STAT3 or SHP-1 activity in experiments (d) and (e). d Effects of SC-2001 on p-STAT3 activity in MDA-MB-468 xenograft tumors. Activity of p-STAT3 was measured by p-STAT3-specific ELISA kit (Cell signaling, Danvers, MA, USA). Columns, mean; bars, SD (n = 6). *p < 0.05. e The activity of SHP-1 in MDA-MB-468 tumors. Columns, mean; bars, SD (n = 6). *p < 0.05
Expression of RFX-1, SHP-1, and p-STAT3 in breast tumor tissue from breast cancer patients
To explore the expression of RFX-1, SHP-1, and p-STAT3 in clinical samples from breast cancer patients, we further examined expressions of these molecules in tumor tissue from 110 patients with breast cancers (including 62 [56.4 %] of TNBC, 42 [38.2 %] of HER-2 positive breast cancers, and 6 [5.4 %] others). Figure 6 shows a representative tumor sample showing RFX-1, SHP-1, p-STAT3 expressions under the same magnification power (200×) comparing breast cancer cells (circled area) with adjacent normal breast cells (area marked with stars). As shown in Fig. 6a, RFX-1 showed weak nuclear expression in breast cancer cells but strong nuclear staining in adjacent normal breast cells (Fig. 6a, upper). SHP-1, transcriptionally regulated by RFX-1, showed similar weak nuclear expression in breast cancer cells but strong nuclear staining in adjacent normal breast cells (Fig. 6a, middle). Conversely, p-STAT3, which can be negatively regulated by SHP-1, showed prominent nuclear staining in breast cancer cells but weak nuclear expression in adjacent normal breast cells (Fig. 6a, lower). Figure 6b the reveals representative reciprocal RFX-1 expression (nuclear staining) and SHP-1 expression (cytoplasmic staining) in breast cancer cells (sample#1 and #2). We found a positive correlation of RFX-1 and SHP-1 expressions in breast cancer cells from tumor samples of the 110 patients with breast cancers (Fig. 6c). Using linear regression model, the correlation coefficient (R 2) was 0.68 (p = 0.006). More studies are necessary to further define the roles of RFX-1, SHP-1, and p-STAT3 and their biological significance in breast cancers.

Immunohistochemical stain for RFX-1, SHP-1, and p-STAT3 in clinical breast tumor samples. a Representative immunohistochemical patterns showing weak nuclear expression for RFX-1 in breast cancer cells and strong nuclear expression for RFX-1 in normal breast cells (upper). Weak nuclear expression for SHP-1 in breast cancer cells and strong nuclear expression for SHP-1 in normal breast cells (middle). Strong nuclear expression for p-STAT3 in breast cancer cells and weak nuclear expression for p-STAT3 in normal breast cells (lower). Asterisk indicates area of cancer cells, circled area indicates adjacent normal breast glands. All pictures were taken under magnification power ×200. Summary of picture findings was shown. B. Representative reciprocal expressions of RFX-1 and SHP-1 in breast cancer cells (sample#1 and #2). c Positive correlation of RFX-1 and SHP-1 expressions in breast cancer cells from tumor samples of 110 patients with breast cancers. Left, linear regression model, the correlation coefficient (R 2) was 0.68. (p = 0.006). Right, histological score (H score) of RFX-1, SHP-1, and p-STAT3 expressions
Discussion
In this study, we demonstrated that SC-2001, an obatoclax derivative, had potent antitumor activity that occurred through SHP-1-dependent STAT3 inactivation. SC-2001 inhibited tumor cell growth and induced apoptosis in both a dose- and time-dependent manner in breast cancer cells including TNBC cells (Fig. 1). The apoptotic effects elicited by SC-2001 were accompanied by decreases in levels of p-STAT3 as well as the downstream apoptotic signaling molecules including anti-apoptotic Bcl-2 family protein Mcl-1, survivin, and cell cycle regulator cyclin D1 (Fig. 2a). SC-2001 downregulated the downstream effectors of STAT3 by reducing transcriptional activation activity of STAT3 (Fig. 2b). Moreover, SC-2001 increased the expression and also the activity of SHP-1, a negative regulator of STAT3, in a time-dependent manner (Fig. 3a–c). The enhanced SHP-1 expression by SC-2001 was associated with the p-STAT3 downregulation and was modulated directly by upregulating SHP-1 transcription by RFX-1 (Fig. 4). Collectively, our data reveal that STAT3, SHP-1 as well as RFX-1 have important roles in mediating SC-2001-induced cell death in TNBC cells.
STAT3 is associated with cell proliferation, survival, and carcinogenesis. Numerous studies have reported that STAT3 is constitutively phosphorylated in a wide variety of cancers through the increased activities of positive regulators, such as the interleukin-6 (IL-6) cytokines, receptor tyrosine kinases EGFR and VEGFR, and non-receptor tyrosine kinases such as Src [32]; and decreased expression of negative regulators, such as the SOCS proteins [33, 34] and tyrosine phosphatases including SHP-1, SHP-2, and PTP-1B [35]. IL-6 is frequently co-overexpressed with its receptor in breast cancer cells and activates JAK/STAT signaling through an autocrine signaling network [36]. Moreover, prolonged upregulation of p-STAT3 has been associated with TNBC [37, 38], in part through autocrine expression of the IL-6 [39–42]. In addition, epigenetic silencing of the negative regulators of JAK/STATs signaling is frequently observed in breast cancers. For example, silencing of the SOCS 1 gene has been reported in 9 % of primary breast cancers [43]. Loss of SHP-1 function may also contribute to the activation of JAK or STAT proteins in cancer, and thus SHP-1 has been implicated as a tumor suppressor [44]. These reports together with our present finding indicate that targeting STAT3 signaling may have a promising therapeutic potential in the treatment of TNBC and other cancers.
SC-2001 was originally derived from obatoclax as a Bcl-2 family inhibitor [30]. Structurally, obatoclax is a synthetic indole bipyrrol derivative. SC-2001 differs from obatoclax at the 5-position of indole structure, where hydrogen is substituted by bromine [30]. Obatoclax is a BH3 mimetic that is designed to occupy the hydrophobic cleft within the BH3-binding groove of Bcl-2 family proteins, disrupt the interaction of Bcl-2 family proteins, and thus induce apoptosis [45]. Our previous study showed that SC-2001 inhibited the protein–protein interactions between Mcl-1 and Bak similar to obatoclax [29]. Here we provide evidence that SC-2001 induces apoptosis in TNBC cells by modulating SHP-1/STAT3 signaling. Currently, there are no specific therapeutic targets for TNBC. Our results support the notion that STAT3 may be a suitable target for TNBC therapy.
The finding that SC-2001 acts as a SHP-1 activator and subsequently targets STAT3 downregulation provides new insight into TNBC drug discovery strategy. Previously, we demonstrated that sorafenib and the derivatives inhibit p-STAT3 by increasing SHP-1 activity without affecting SHP-1 expression [28, 46, 47]. In addition, several compounds including β-escin, gamma-tocotrienol, acety-11-keto-beta-boswellic acid, butein (3,4,2′,4′-tetrahydroxychalcone), and betulinnic acid have also been shown to enhance SHP-1 activity which accounts for the downregulation of STAT3 activity and mediates their anti-cancer effects on various kinds of cancer cells [20, 21, 48, 49]. Taken together, the common mechanism employed by these structurally distinct agents in various cancer cells highlights a possibility that targeting the activity or interaction of phosphatases and oncokinases could be a promising anti-TNBC or anti-cancer strategy.
In this study, we also demonstrated an indispensable role of RFX-1, one of the major transcription factors of SHP-1, in SC-2001-induced anti-cancer effects (Fig. 5). SC-2001 increased RFX-1 expression and subsequently activated SHP-1 transcription, both in vitro (Fig. 5) and in TNBC xenografts (Fig. 6c, e). RFX-1 belongs to the regulatory factor X gene family, which is known to regulate major histocompatibility complex class II gene expression [50]. The role of RFX-1 in cancer remains unclear. Previous reports have suggested potential tumor-suppressive role of RFX-1 by negatively regulating cancer cell proliferation, including breast cancer cells [31, 51–55]. In addition, lower expression of RFX-1 has been found in human glioma tissues or cell lines, compared with that in normal brain tissue [54]. Similarly, the expression of RFX-1 has been reported to be decreased in esophageal adenocarcinoma, as compared with that in precancerous Barrett’s esophagus epithelium [56]. Interestingly, we have shown here that a representative breast tumor tissue has reciprocal expression of RFX-1, SHP-1, and p-STAT3 in cancer cells and adjacent non-cancer breast tissue (Fig. 4). Large immunohistochemistry-based studies are needed to address the correlation of RFX-1 and SHP-1, as well as p-STAT3 expression in heterogeneous breast cancer subtypes. The current results notwithstanding, the detailed mechanisms by which SC-2001 enhance RFX-1 expression, remain to be elucidated. Further studies are necessary to evaluate the role of RFX-1 in breast cancer carcinogenesis and the mechanism by which RFX-1 is modulated in breast cancer cells.
Conclusions
SC-2001 shows a beneficial apoptosis-inducing effect in breast cancer cells including TNBC cells through a mechanism involving RFX-1/SHP-1-dependent p-STAT3 downregulation. This study indicates that targeting STAT3 signaling pathway may be a useful approach for the development of targeted agents for anti-breast cancer, especially anti-TNBC treatments.
Abbreviations
- HER2:
-
Human epidermal growth factor receptor 2
- TNBC:
-
Triple-negative breast cancer
- JAK:
-
Janus kinase
- ALK:
-
Anaplastic lymphoma kinase
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- FLT-3:
-
FMS-like tyrosine kinase 3
- MEK:
-
Mitogen-activated protein kinase kinase
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- STAT3:
-
Signal transducers and activators of transcription 3
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- IL-6:
-
Interleukin-6
- PARP:
-
Poly ADP-ribose polymerase
- DMSO:
-
Dimethyl sulfoxide
- PTP:
-
Protein tyrosine phosphatase
- SHP:
-
Src homology 2-domain-containing tyrosine phosphatase
- VEGFR:
-
Vascular endothelial growth factor receptor
- PDGFR:
-
Platelet-derived growth factor receptor
- EGF:
-
Epidermal growth factor
- ERK1/2:
-
Extracellular signal-regulated protein kinases 1 and 2
- FGFR:
-
Fibroblast growth factor receptor
- RFX:
-
Regulatory factor X
References
Alvarez RH, Valero V, Hortobagyi GN (2010) Emerging targeted therapies for breast cancer. J Clin Oncol 28(20):3366–3379
Boyle P (2012) Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol 23 Suppl 6:vi7–vi12
Elias AD (2010) Triple-negative breast cancer: a short review. Am J Clin Oncol 33(6):637–645
Duffy MJ, McGowan PM, Crown J (2012) Targeted therapy for triple-negative breast cancer: where are we? Int J Cancer 131(11):2471–2477
Fulda S (2009) Tumor resistance to apoptosis. Int J Cancer 124(3):511–515
Oakes SR, Vaillant F, Lim E, Lee L, Breslin K, Feleppa F, Deb S, Ritchie ME, Takano E, Ward T, Fox SB, Generali D, Smyth GK, Strasser A, Huang DC, Visvader JE, Lindeman GJ (2012) Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc Natl Acad Sci USA 109(8):2766–2771
Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, Bornmann W, Kantarjian H, Viallet J, Samudio I, Andreeff M (2008) Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res 68(9):3413–3420
Martin AP, Mitchell C, Rahmani M, Nephew KP, Grant S, Dent P (2009) Inhibition of MCL-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol Ther 8(21):2084–2096
Tang Y, Hamed HA, Cruickshanks N, Fisher PB, Grant S, Dent P (2012) Obatoclax and lapatinib interact to induce toxic autophagy through NOXA. Mol Pharmacol 81(4):527–540
Witters LM, Witkoski A, Planas-Silva MD, Berger M, Viallet J, Lipton A (2007) Synergistic inhibition of breast cancer cell lines with a dual inhibitor of EGFR-HER-2/neu and a Bcl-2 inhibitor. Oncol Rep 17(2):465–469
Diaz N, Minton S, Cox C, Bowman T, Gritsko T, Garcia R, Eweis I, Wloch M, Livingston S, Seijo E, Cantor A, Lee JH, Beam CA, Sullivan D, Jove R, Muro-Cacho CA (2006) Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res 12(1):20–28
Germain D, Frank DA (2007) Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res 13(19):5665–5669
Bromberg J (2000) Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res 2(2):86–90
Marzo I, Naval J (2008) Bcl-2 family members as molecular targets in cancer therapy. Biochem Pharmacol 76(8):939–946
Ling X, Arlinghaus RB (2005) Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res 65(7):2532–2536
Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, Pandit B, Ihnat MA, Shenoy SS, Kulp S, Li PK, Li C, Fuchs J, Lin J (2010) Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res 70(6):2445–2454
Gu F, Dube N, Kim JW, Cheng A, Ibarra-Sanchez Mde J, Tremblay ML, Boisclair YR (2003) Protein tyrosine phosphatase 1B attenuates growth hormone-mediated JAK2-STAT signaling. Mol Cell Biol 23(11):3753–3762
Insabato L, Amelio I, Quarto M, Zannetti A, Tolino F, de Mauro G, Cerchia L, Riccio P, Baumhoer D, Condorelli G, Terracciano L, de Franciscis V (2009) Elevated expression of the tyrosine phosphatase SHP-1 defines a subset of high-grade breast tumors. Oncology 77(6):378–384
Esposito N, Colavita I, Quintarelli C, Sica AR, Peluso AL, Luciano L, Picardi M, Del Vecchio L, Buonomo T, Hughes TP, White D, Radich JP, Russo D, Branford S, Saglio G, Melo JV, Martinelli R, Ruoppolo M, Kalebic T, Martinelli G, Pane F (2011) SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome-positive cells derived from patients with chronic myeloid leukemia. Blood 118(13):3634–3644
Pandey MK, Sung B, Aggarwal BB (2010) Betulinic acid suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase SHP-1 in human multiple myeloma cells. Int J Cancer 127(2):282–292
Kunnumakkara AB, Nair AS, Sung B, Pandey MK, Aggarwal BB (2009) Boswellic acid blocks signal transducers and activators of transcription 3 signaling, proliferation, and survival of multiple myeloma via the protein tyrosine phosphatase SHP-1. Mol Cancer Res 7(1):118–128
Prasad S, Pandey MK, Yadav VR, Aggarwal BB (2011) Gambogic acid inhibits STAT3 phosphorylation through activation of protein tyrosine phosphatase SHP-1: potential role in proliferation and apoptosis. Cancer Prev Res (Phila) 4(7):1084–1094
Phromnoi K, Prasad S, Gupta SC, Kannappan R, Reuter S, Limtrakul P, Aggarwal BB (2011) Dihydroxypentamethoxyflavone down-regulates constitutive and inducible signal transducers and activators of transcription-3 through the induction of tyrosine phosphatase SHP-1. Mol Pharmacol 80(5):889–899
Pandey MK, Sung B, Ahn KS, Aggarwal BB (2009) Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol Pharmacol 75(3):525–533
Kang SH, Jeong SJ, Kim SH, Kim JH, Jung JH, Koh W, Kim DK, Chen CY (2012) Icariside II induces apoptosis in U937 acute myeloid leukemia cells: role of inactivation of STAT3-related signaling. PLoS ONE 7(4):e28706
Sandur SK, Pandey MK, Sung B, Aggarwal BB (2010) 5-Hydroxy-2-methyl-1,4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: potential role in chemosensitization. Mol Cancer Res 8(1):107–118
Liu CY, Tseng LM, Su JC, Chang KC, Chu PY, Tai WT, Shiau CW, Chen KF (2013) Novel sorafenib analogues induce apoptosis through SHP-1 dependent STAT3 inactivation in human breast cancer cells. Breast Cancer Res 15(4):R63
Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ, Chen KF (2011) Signal transducer and activator of transcription 3 is a major kinase-independent target of sorafenib in hepatocellular carcinoma. J Hepatol 55(5):1041–1048
Chen KF, Su JC, Liu CY, Huang JW, Chen KC, Chen WL, Tai WT, Shiau CW (2012) A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett 321(1):27–35
Su JC, Chen KF, Chen WL, Liu CY, Huang JW, Tai WT, Chen PJ, Kim I, Shiau CW (2012) Synthesis and biological activity of obatoclax derivatives as novel and potent SHP-1 agonists. Eur J Med Chem 56:127–133
Amin S, Kumar A, Nilchi L, Wright K, Kozlowski M (2011) Breast cancer cells proliferation is regulated by tyrosine phosphatase SHP1 through c-jun N-terminal kinase and cooperative induction of RFX-1 and AP-4 transcription factors. Mol Cancer Res 9(8):1112–1125
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7(1):41–51
Larsen L, Ropke C (2002) Suppressors of cytokine signalling: SOCS. APMIS 110(12):833–844
Gao Y, Cimica V, Reich NC (2012) Suppressor of cytokine signaling 3 inhibits breast tumor kinase activation of STAT3. J Biol Chem 287(25):20904–20912
Yu H, Jove R (2004) The STATs of cancer—new molecular targets come of age. Nat Rev Cancer 4(2):97–105
Conze D, Weiss L, Regen PS, Bhushan A, Weaver D, Johnson P, Rincon M (2001) Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res 61(24):8851–8858
Berclaz G, Altermatt HJ, Rohrbach V, Siragusa A, Dreher E, Smith PD (2001) EGFR dependent expression of STAT3 (but not STAT1) in breast cancer. Int J Oncol 19(6):1155–1160
Yeh YT, Ou-Yang F, Chen IF, Yang SF, Wang YY, Chuang HY, Su JH, Hou MF, Yuan SS (2006) STAT3 ser727 phosphorylation and its association with negative estrogen receptor status in breast infiltrating ductal carcinoma. Int J Cancer 118(12):2943–2947
Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, Bornmann W, Bromberg JF (2007) Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res 9(3):R32
Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, Chieco P, Bonafe M (2007) IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest 117(12):3988–4002
Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28(33):2940–2947
Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA, Polyak K (2011) The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24− stem cell-like breast cancer cells in human tumors. J Clin Invest 121(7):2723–2735
Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, Campbell IG, Visvader JE (2004) Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 23(46):7726–7733
Wu C, Sun M, Liu L, Zhou GW (2003) The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene 306:1–12
Ni Chonghaile T, Letai A (2008) Mimicking the BH3 domain to kill cancer cells. Oncogene 27(Suppl 1):S149–S157
Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, Li PK, Chen PJ, Cheng AL (2010) Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res 16(21):5189–5199
Chen KF, Chen HL, Shiau CW, Liu CY, Chu PY, Tai WT, Ichikawa K, Chen PJ, Cheng AL (2013) Sorafenib and its derivative SC-49 sensitize hepatocellular carcinoma cells to CS-1008, a humanized anti-TNFRSF10B (DR5) antibody. Br J Pharmacol 168(3):658–672
Tan SM, Li F, Rajendran P, Kumar AP, Hui KM, Sethi G (2010) Identification of beta-escin as a novel inhibitor of signal transducer and activator of transcription 3/Janus-activated kinase 2 signaling pathway that suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells. J Pharmacol Exp Ther 334(1):285–293
Rajendran P, Li F, Manu KA, Shanmugam MK, Loo SY, Kumar AP, Sethi G (2011) gamma-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br J Pharmacol 163(2):283–298
Gajiwala KS, Chen H, Cornille F, Roques BP, Reith W, Mach B, Burley SK (2000) Structure of the winged-helix protein hRFX1 reveals a new mode of DNA binding. Nature 403(6772):916–921
Chen L, Smith L, Johnson MR, Wang K, Diasio RB, Smith JB (2000) Activation of protein kinase C induces nuclear translocation of RFX1 and down-regulates c-myc via an intron 1× box in undifferentiated leukemia HL-60 cells. J Biol Chem 275(41):32227–32233
Liu M, Lee BH, Mathews MB (1999) Involvement of RFX1 protein in the regulation of the human proliferating cell nuclear antigen promoter. J Biol Chem 274(22):15433–15439
Hsu YC, Liao WC, Kao CY, Chiu IM (2010) Regulation of FGF1 gene promoter through transcription factor RFX1. J Biol Chem 285(18):13885–13895
Ohashi Y, Ueda M, Kawase T, Kawakami Y, Toda M (2004) Identification of an epigenetically silenced gene, RFX1, in human glioma cells using restriction landmark genomic scanning. Oncogene 23(47):7772–7779
Feng C, Zuo Z (2012) Regulatory factor X1-induced down-regulation of transforming growth factor beta2 transcription in human neuroblastoma cells. J Biol Chem 287(27):22730–22739
Watts JA, Zhang C, Klein-Szanto AJ, Kormish JD, Fu J, Zhang MQ, Zaret KS (2011) Study of FoxA pioneer factor at silent genes reveals Rfx-repressed enhancer at Cdx2 and a potential indicator of esophageal adenocarcinoma development. PLoS Genet 7(9):e1002277
Acknowledgments
This research was supported by grants from the Taiwan Clinical Oncology Research Foundation and Yen Tjing Ling Medical Foundation; MOST103-2325-B-075-002, NSC101-2325-B-002-032, NSC102-2325-B-075-003, NSC101-2325-B-075-006, NSC100-2325-B-010-007, and NSC101-2325-B-010-007 from the Ministry of Science and Technology, Taiwan; V102A-005, V101B-003, V102B-011, V103C-141 from Taipei Veterans General Hospital; TVGH-NTUH Joint Research Program VN103-08 from Taipei Veterans General Hospital and National Taiwan University Hospital, and MOHW103-TD-B-111-02 from the Center of Excellence for Cancer Research at Taipei Veterans General, the Ministry of Health and Welfare, Executive Yuan, Taiwan.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This study was approved by the ethics committee of the Institutional Review Board of Taipei Veterans General Hospital. All experiments comply with the current laws of Taiwan. Informed consent was obtained from all sample donors at time of their donation in accordance with the Declaration of Helsinki. Animal experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Chun-Yu Liu and Jung-Chen Su contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
The chemical structure of obatoclax and SC-2001. Supplementary material 1 (TIFF 477 kb)
Supplementary Fig. 2
1st, SHP-1 activity of MDA-MB-468 cells exposed to SHP-1 inhibitor (50 and 100 μM) for 24 h. 2nd, Protein expression of SHP-1, p-STAT3, and STAT3 in MDA-MB-468 cells after treatment of SHP-1 inhibitor (50 and 100 μM) and vanadate (25 μM) for 24 h. Beta-actin was served as loading control. 3rd, cell viability of MDA-MB-468 cells exposed to vanadate or SHP-1 inhibitor for 24 h, respectively. The cell viability was measured by MTT assay. Supplementary material 2 (TIFF 776 kb)
Supplementary Fig. 3
RFX-1 signaling cascade is the cause of SC-2001-mediated cell death. Left, mRNA levels of RFX-1 in three TNBC cells after treatment of SC-2001 for 0, 9, and 18 h. Right, cell viability of three TNBC cells after treatment of SC-2001 for 0, 9, and 18 h. The cell viability was measured by MTT assay. Supplementary material 3 (TIFF 671 kb)
Rights and permissions
About this article

Cite this article
Liu, CY., Su, JC., Ni, MH. et al. Obatoclax analog SC-2001 inhibits STAT3 phosphorylation through enhancing SHP-1 expression and induces apoptosis in human breast cancer cells. Breast Cancer Res Treat 146, 71–84 (2014). https://doi.org/10.1007/s10549-014-3000-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-014-3000-0