
Abstract
Nicotine, a main addictive compound in tobacco smoke, has been linked to promotion and progression of lung, head and neck, pancreatic, and breast cancers, but the detailed mechanisms of cancer progression remain elusive. Here, we show that nicotine induces the expression of galectin-3 (an anti-apoptotic β-galactoside-binding lectin) in breast cancer cell line and in primary tumors from breast cancer patients. Nicotine-induced up regulation of galectin-3 is due to an increased expression of α9 isoform of nicotinic acetylcholine receptor (α9nAChR), which activates transcription factor STAT3 that in turn, physically binds to galectin-3 (LGALS3) promoter and induces transcription of galectin-3. Intracellular galectin-3 increased mitochondrial integrity and suppressed chemotherapeutic-induced apoptosis of breast cancer cell. Moreover, nicotine-induced enrichment of side population cells with cancer stem cell-like properties was modulated by galectin-3 expression and could be significantly reduced by transient knock down of LGALS3 and its upstream signaling molecules STAT3 and α9nAChR. Thus, galectin-3 or its upstream signaling molecule STAT3 or α9nAChR could be a potential target to prevent nicotine-induced chemoresistance in breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
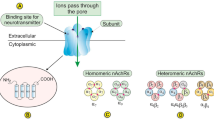
Smoking is estimated to account for approximately 4–5 million deaths worldwide and approximately 443,000 deaths each year in the United States alone [1, 2]. Nicotine, the main addictive component of tobacco smoke, has been shown to promote cell proliferation, angiogenesis, and epithelial–mesenchymal transition (EMT), leading to enhanced tumor growth and metastasis [2–4]. Moreover, nicotine has been shown to abrogate the beneficial effect of chemotherapeutic drugs by inhibiting apoptosis in lung cancer [5]. The effects of nicotine are usually mediated through the nicotinic acetylcholine receptors (nAChRs) that are expressed on a variety of neuronal and non-neuronal cells [2]. nAChRs are complex of five subunits (comprising of α2–α10 and β2–β4) forming homopentamers (neuronal nAChRs) or heteropentamers (muscle type nAChRs) to form a central ion channel. Nicotine mimics acetylcholine by binding as an agonist to α subunit of nAChRs and α7nAChR is found the major stimulator of cancer development and progression in vivo in many cancers [2]. In breast cancer cells, at least four different subunits (α5, α7, α9, and β4) of nAChRs are known, but nicotine exposure increases mostly the expression of α9nAChR [6].
Signal transducer and activator of transcription 3 (STAT3) is a major transcription factor for mediating cytokine responses [7]. In response to cytokines and growth factors, STAT3 is phosphorylated at tyrosine705 by receptor-associated Janus tyrosine kinases and then forms homo- or heterodimers that translocate to the cell nucleus, where it drives transcription by binding to specific sequences. Inactivation of STAT3 in experimental animals leads to the development of several pathologies including embryonic lethality [7]. STAT3 is constitutively activated in human tumor cell lines and primary tumors and its constitutive activation commonly suggests poor prognosis [8, 9]. Recent studies suggest that nicotine/cigarette smoke could activate STAT3 in various pathological models including cancer [10–12].
Galectin-3, a member of at least fifteen β-galactoside-binding soluble lectins family is involved in tumor cell adhesion, angiogenesis, cancer progression, and metastasis [13–16]. Galectin-3 expression in gastric, liver, lung, bladder, and head and neck cancers was significantly increased compared to the normal tissues, and correlated with the progression of clinical stages and formation of metastases [17–20]. Interestingly, a change in cellular localization of galectin-3 was observed during progression of various cancers. Down-regulation of surface galectin-3 expression in colon and tongue cancers, with an increased cytoplasmic expression of galectin-3 at more advanced stages was reported [21, 22]. Several studies suggest that cytoplasmic galectin-3 inhibits apoptosis similar to Bcl-2 [23–25]. Resistance to apoptosis is not only essential for cancer cell survival but also for tumor progression. Conversely, secreted galectin-3 from tumor cell induces T cell apoptosis implicating a possible role in immune escape mechanism during tumor progression [16, 23]. Several recent studies highlighted the clinical and biological significance of increased galectin-3 expression in apoptosis resistance in cancer cells in connection to targeted cancer therapies and also documented therapeutic effects of synthetic carbohydrate-based small molecule inhibitors of galectin-3 [26–29]. However, studies examining the relevance of galectin-3 to nicotine and STAT3 or the possible roles of nAChR in the regulation of galectin-3 have not been reported to date.
Here we present evidence that nicotine promotes galectin-3 expression in breast cancer cells. Nicotine activated STAT3 through α9nAChR, which then promoted galectin-3 expression in breast cancer cells. Overexpression of galectin-3 promoted chemoresistance through a nicotine dependent anti-apoptosis and an enrichment of side populations with cancer stem cell-like properties.
Methods
Cell culture and cell transfection
The breast cancer cell line MCF-7 was obtained from (ATCC) and cultured in Dulbecco’s modified Eagle’s medium with 10 % fetal bovine serum (FBS) and antibiotics. Cells were maintained in a humidified incubator at 37 °C in the presence of 5 % CO2. The transfection of cells was performed with TurboFect (Thermo Scientific) according to the manufacturer’s instructions.
Antibodies and reagents
Antibodies against phospho-(Tyr705)-STAT3 and STAT3 were purchased from Cell signaling Technologies. Antibodies against TWIST1, α9-nicotinic acetylcholine receptor (α9nAChR), endo G, and galectin-3 were obtained from Aviva Systems Biology. Beta actin antibody was obtained from Sigma. Nicotine and staurosporine were purchased from Sigma. Mitotracker (mitochondrion selective probe) was obtained from Invitrogen. SiRNA of galectin-3 (Duplex no. 2, siSTABLE) beginning at nt 518, 5′-GCAAUACAAAGCUGGAUAAdTdT-3′ (sense), 5′-P UUAUCCAGCUUUGUAUUGCdTdT-3′ (antisense) was purchased from Dharmacon Research (Lafayette, CO). Galectin-3 MISSION shRNA Lentiviral Transduction Particles with target sequence CCGGGCTCACTTGTTGCAGTACAATCTCGAGATTGTACTGCAACAAGTGAGCTTTTT (Cat No. SHCLNV-NM_002306) was purchased from Sigma. Negative Control Mission shRNA Transduction Particles (Cat No. SHC002V) was obtained from Sigma. Human STAT3-specific shRNAs (shRNA1-STAT3, shRNA2-STAT3, shRNA3-STAT3) were synthesized from pLKO.1 vector as previously described [30]. Target sequences of shRNA1-STAT3 (ATCTCCTGACCTTATGATCCG) are located at the 3′UTR starting at nucleotide 3170 (Genbank accession no. NM003150). Target sequences of shRNA2-STAT3 (TTCTTGGGATTGTTGGTCAGC) and shRNA3-STAT3 (TTGATTCTTCGTAGATTGTGC) are located at the coding region starting at nucleotides 1663 and 728, respectively. Vector containing scrambled shRNA was used as a negative control. Corresponding negative shRNA for STAT3 is the empty vector. Mission endo-ribonuclease prepared siRNAs (esiRNAs) of α9nAChR (CHRNA9) are a heterogeneous mixture of siRNAs synthesized by in vitro transcription of a 425 bp gene specific dsRNA, followed by enzymatic digestion using RNaseIII (Cat No. EHU008481, Sigma). Mission esiRNAs of TWIST1 are a heterogeneous mixture of siRNAs synthesized by in vitro transcription of a 459 bp gene specific dsRNA, followed by enzymatic digestion using RNaseIII (Cat No. EHU151571, Sigma). Negative control siRNA (Cat No. SIC001) was purchased from Sigma. All gene-specific siRNAs/shRNAs knocked down the expression of the corresponding genes as validated on either RT-PCR or Western blot.
Western blot
Western blot analyses were performed as previously described [31]. Briefly the cells were lysed using Mammalian Cell Lysis kit (Sigma) that includes 250 mM Tris–HCl, pH 7.5, 5 mM EDTA, 750 mM NaCl, 0.5 % SDS, 2.5 % Deoxycholic acid, 5 % Igepal, and Protease inhibitor cocktail. After centrifugation, the clear supernatant was separated by SDS–PAGE and immunoblotted with appropriate primary antibodies as indicated in the figure legends. Specific bands were detected with HRP conjugated anti-rabbit IgG (Cell Signaling) and Enhanced Chemiluminescence reagent (Thermo Scientific). Original gel images are presented and no editing of gel images was performed.
For a Western blot analysis of the breast cancer tissues (smoker and non-smoker), paraffin embedded tissue sections were deparaffinized with xylene for 10 min followed by hydration using 100, 95, and 70 % alcohol (5 min each), and finally washing with distilled water. Tissue samples were scraped using an aseptic needle followed by extraction of protein in 2× Laemmli sample buffer. Sample was boiled in 100 °C for 10 min, centrifuged, and the clear supernatant was subjected to Western blotting.
RNA isolation and gene expression on quantitative real time polymerase chain reaction (qRT-PCR) and PCR micoarray
Total RNA was extracted using the All-in-One Purification kit (Norgen Biotek Corporation) and quantified spectrophotometrically using the NanoDrop 2000 spectrophotometer (Thermo Scientific). The first strand cDNA synthesis was carried out using RT2 First Strand Kit (SA Biosciences) using 4 μg of total RNA and oligo (dT) primers and PCR was performed with gene specific primers (α9nAChR: forward 5′-GACTGTTCGTTTCCCAGATGG-3′ and reverse 5′-ACGAAGTTGGGAGCCGACATCA-3′; galectin-3: forward 5′-CACGGTGAAGCCCAATGCA-3′ and reverse 5′-GTGAGCATCATTCACTGCAACC-3′). Primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (used as housekeeping gene control) were: forward 5′-TTGCCATCAATGACCCCTTCA-3′ and reverse 5′-CGCCCCACTTGATTTTGGA-3′. The expression of each gene was quantified by using the comparative Ct (ΔΔCt) method as described in the Assays-on-Demand Users Manual (Applied Biosystems). The fold values (X) were calculated using the formula: X = 2(−ΔΔCt), where the data for the sample and sham-treated cell (here, the calibrator being the sham-treated cells) were first normalized against variations of sample quality and quantity. The ΔΔCt was determined using the formula: ΔΔCt = ΔC(t) sample −ΔC(t)calibrator, where ΔC(t) sample = C(t) target gene of sample − C(t) reference, and ΔC(t)calibrator = C(t) target gene of calibrator − C(t) reference. The expression of the target genes was normalized to GAPDH and relative to the calibrator = 2(−ΔΔCt). Gene expression analysis was also performed using Cancer Pathway Finder PCR array (SA Biosciences, Qiagen) and calculated according to the manufacturer’s instructions.
Scratch–wound healing assay
Scratch–wound healing assay was performed as previously described with some modifications [32]. Biriefly, MCF-7 cells were seeded in a 6-well plate and cultured overnight to form confluent monolayers. The cell monolayer was replaced with serum-free medium for overnight and a wound was introduced into the monolayer using a pipette tip. After washing with serum-free medium, the denuded area was photographed (0 h). Cells were then cultured in serum-free medium in absence or in the presence of nicotine (100 μM) for 72 h and following washing with 1× phosphate-buffered saline [(PBS) 10 mM phosphate, 140 mM NaCl, pH 7.5] wound images were photographed. The numbers of migrated cells into denuded area of three different samples were counted. The results were expressed as mean cell number ± SD.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed as previously described with some modifications [33]. Briefly, soluble chromatin was prepared from a total of 2 × 107 MCF-7 cells that were untreated or treated with nicotine (100 μM). The chromatin solution was diluted tenfold with ChIP dilution buffer (1.1 % Triton X-100, 1.2 mM EDTA, 167 mM NaCl, 16.7 mM Tris–HCl, pH 8.1, 0.01 % SDS, inhibitors of protease and phosphatase), pre cleared with protein A beads that were pre-blocked with 2 μg of sheared salmon sperm DNA and pre-immune serum. The pre-cleared chromatin solution was immunoprecipitated with anti-phospho-(Tyr705)-STAT3 antibody (Cell Signaling). Following multiple washes, the protein-DNA complex was eluted from the beads with 1 % SDS and 0.1 M NaHCO3. After reversing crosslinks, protein and RNA were removed by digestion with 10 μg proteinase K and 10 μg of RNAase A at 42 °C for 3 h, respectively. Purified DNA was subjected to PCR with primers specific for the indicated gene promoters. The sequences of the PCR primers used for TWIST1 are forward, 5′-GCCAGGTCGTTTTTGAATGG-3′and reverse, 5′-CGTGCAGGCGGAAAGTTTGG-3′, and for galectin-3 are forward 5-GGAGAGGGCGGGGGATAG-3″ and reverse 5″-ACGCCCTCTCCCCTACCC-3′.
Immunohistochemical staining
Tissue slides of breast cancer patients (smoker and non-smoker) with varying stages were purchased from Pathology Biorepository and Research Core of the University of Maryland Greenebaum Cancer Center under approved Institutional Review Board protocol (see Table 1 for details). Galectin-3, α9nAChR, (Tyr705)-phospho-STAT3, and TWIST1 were detected using specific antibodies on paraffin-embedded sections of the tumor as previously described [31]. Briefly, sections were deparaffinized with xylene followed by hydration with graded concentrations of ethanol and then with distilled water. Samples were subjected to antigen retrieval followed by washing with PBS and incubation in 3 % hydrogen peroxide to inhibit endogenous peroxidase. The tissue sections were blocked with 1 % bovine serum albumin (BSA) and incubated with the indicated primary antibody (1:100) in BSA for 1 h. After washing, the sections were incubated with HRP conjugated secondary antibody for 1 h and positive signals were visualized with the diaminobenzidine (DAB) substrate. The number of cells showing positive staining of galectin-3, α9nAChR, TWIST1 and phospho-(Tyr705)-STAT3 were scored independently and blindly in 3 different area of each slide and then data were expressed as mean ± SD, where *** p < 0.001.
Side population (SP) study
SP cells are analyzed by flow cytometer equipped with violet laser diode (VLD) and DyeCycle Violet (DCV) fluorescence is measured through blue and red band pass filter. Dual-parametric analysis of Forward scatter (FSC) versus side scatter (SSC) is the primary gate used to identify cells in the appropriate size range, and to eliminate debris. Cells falling in the primary gate are subsequently evaluated by analysis of SSC height versus SSC width, followed by FSC height versus FSC width, for doublet discrimination. Finally, the live, singlet cells are displayed in a dual parametric presentation of DCV red fluorescence (x axis) versus DCV blue fluorescence (y axis), and the location of the SP is determined based upon loss of the putative SP in a cohort population pre-incubated with verapamil. DyeCycle Violet blue and red fluorescence are plotted allowing visualization of low fluorescence “tail” of cells (called SP cells, represented in side pink box of the plot) relative to the much larger bulk of DyeCycle Violet stained cells. Side population (SP) of breast cancer cells has recently been shown to harbor stem cell-like properties with resistance to apoptosis [34–36]. For side population study, MCF-7 cells were removed from the culture dish with trypsin and EDTA (GIBCO BRL), washed, suspended at 106 cells per ml in DMEM containing 2 % Fetal calf serum (FCS) (staining medium), and pre-incubated in a 1.5-ml Eppendorf tube at 37 °C for 10 min. Cells were then labeled in the same medium at 37 °C for 30 min with 5 μM Vybrant DyeCycle Violet Stain (Molecular Probes), either alone or in combination with 50 μM verapamil (Sigma) [an inhibitor of ABC transporters [34–36]. Then, 3–5 × 106 cells were analyzed using the Becton–Dickinson FACSVantage fluorescence-activated cell sorter.
Cell viability assays
Cell viability was quantified using commercially available WST-1 colorimetric kit (Roche). Cells were cultured in a clear bottom 96-well plate and the final volume of culture medium in each well was maintained 100 μl. After treating with various agents, 10 μl of WST-1 was added to each well of the plate incubated at 37 °C for 2 h and absorbance at 450 nm was measured using an ELISA plate reader.
Apoptosis assay
Following treatment of MCF-7 cells (approximately 10,000) with siRNA (galectin-3 specific or negative control) for 24 h, cells were incubated with nicotine (100 μM, 48 h) plus staurosporine (STS, 1 μM for last 24 h). One set of cells received only staurosporine (1 μM for last 24 h). Cells were washed with ice cold PBS, stained with annexin-V FITC (Enzo Life Sciences) according to the manufacturer’s instructions and analyzed by flow cytometry using FL1 channel within 30 min.
STS-induced DNA fragmentation of cells in the presence of nicotine alone or nicotine plus siRNA (galectin-3 specific or negative control) was assessed from the determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) using The Cell Death Detection ELISA PLUS kit (Roche Applied Science).
JC-1 staining
The JC-1 staining solution was prepared from the stock (MitoPT™-JC1 Assay kit, Immunochemistry Technologies) by diluting the reagent 1:10 in the culture medium (DMEM). Cells (5 × 105/ml) were plated in 6-well plate and treated with siRNAs, nicotine, and staurosporine, and diluted JC-1 staining solution (200 μl) was added to each well. Cells were grown at 37 °C for an additional 30 min prior to harvesting and spectrofluorimetry (excitation at 480 nm). Healthy cells with functional mitochondria containing red JC-1 aggregates were detected at 590 nm. Apoptotic cells with collapsed mitochondria containing green JC-1 monomers were detected at 527 nm.
Mammosphere assay
Mammosphere culture was performed as previously described [37] with some modifications. Briefly, single-cell suspensions were plated (10,000 cells/well) in 6-well ultra-low attachment plates (Corning) in mammocult medium (Stem cell Technologies) and cultured for 3 days. Mammosphere/Tumorspheres were counted using phase contrast microscope (Axioplane 2 Zeiss microscope fitted with a cooled CCD Olympus model DP70 camera).
Statistics
Data were expressed as mean ± SD unless otherwise mentioned. Comparisons were made between different treatments (analysis of variance) using the software GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA), where an error protecting the multiple comparison procedure, namely Tukey–Kramer multiple comparison tests, was applied for the analysis of significance of all the data.
Results
Nicotine increases apoptosis resistance and cell migration
Nicotine was shown to increase apoptosis resistance and cell migration leading to tumorigenesis in breast cancer cells, particularly MCF-7 [34, 38, 39]. We used staurosporine (STS) (protein kinase inhibitor known to induce mitochondrion-dependent apoptosis) to examine the efficacy of nicotine in promoting resistance to MCF-7 cell death. Treatment of MCF-7 cells with 1 μM STS for 24 h induced significant cell death (69.4 %, p < 0.001). Interestingly, nicotine protected cells from cell death in a dose-dependent manner (8.7, 23.5, and 50 % protection with 1, 10, and 100 μM nicotine, respectively) (Fig 1a). Nicotine alone at up to 100 μM concentration did not show any toxicity on MCF-7 cells (data not shown). Next, we evaluated the role of nicotine on cell migration. As 100 μM nicotine gave most chemoresistance to MCF7 cell without any toxicity on cell viability, we used 100 μM nicotine for most follow-up experiments.

Cell viability and Scratch-wound healing assay. a MCF 7 cells were plated in 96-well plate and treated with staurosporine (1 μM) alone or in combination of varying concentration of nicotine as 100, 10, 1 μM for 24 h followed by cell viability study using WST-1. ***p < 0.001 and *p < 0.05 compared to staurosporine treated cell. b Confluent monolayer of MCF7 cells were replaced with serum-free medium for overnight and a wound was created by dragging a pipette tip through the monolayer. After washing with serum-free medium, wound images were photographed (0 h). Cells were then cultured in serum-free medium in absence or in the presence of nicotine (100 μM) for 72 h and following washing with 1× PBS wound images were photographed. Migratory cells to the denuded area for each of untreated and nicotine-treated cells were counted and expressed as mean ± SD, n = 3. Magnification 5×. c A bar graph showing quantitative data was represented as ± SD. ***p ≤ 0.001
Consistent with previous reports [38, 39], in scratch-wound healing assay nicotine-treated MCF-7 cells (100 μM, 72 h) increased cell migration by almost fourfold (p < 0.001) compared to the untreated control cells (Fig. 1b, c).
Nicotine increases expression of galectin-3
As galectin-3 was involved in tumor cell migration and metastasis [13–16], we evaluated the expression of galectin-3 in breast tissue specimens from cancer patients who smoked or never smoked by immunostaining with anti-galectin-3 antibody [31]. Expression of galectin-3 (gal3) was significantly increased in a stage-dependent manner in both smokers (p < 0.001) and non-smokers (p < 0.05) (Fig. 2a, i, ii). Moreover, galectin-3 content was significantly (p < 0.001) higher in all stages of breast cancer patients who smoked compared to those who never smoked (Fig. 2a, i, ii). This observation was corroborated by a Western blot analysis where expression of galectin-3 in breast cancer tissue specimen (stage III) from smoker was about twofold compared to that from a non-smoker matching tissue specimen (Fig. 2a, iii). However, one cannot conclude that galectin-3 expression is induced by cigarette smoking on the basis of this finding. It is possible that smoking can affect the types of tumors that develop, which in turn are associated with the expression levels of galectin-3. Next, we investigated the effect of nicotine on galectin-3 expression in MCF-7 cells. A qRT-PCR analysis of the expression of LGALS3 mRNA showed a dose-dependent increase (1.7-fold at 1 μM and fivefold at 100 μM of nicotine) (Fig. 2b). The increased expression of galectin-3 protein in nicotine-treated MCF-7 cells was also confirmed by a Western blot analysis (Fig. 2c).

Nicotine-induced galectin-3 expression. a i Representative diagrams showing expression of galectin-3 in various stages of breast cancer tissue specimens from 22 patients (see Table 1 for description) who never smoked and smoked as determined by immunostaining with anti-galectin-3 antibody. Magnification 400×. ii A bar graph showing quantitative expression of galectin-3 for each stage of smoker and non-smoker breast cancer patients (n = 22). Galectin-3 staining of each sample was performed in duplicate or in triplicate. The number of cells showing positive staining of galectin-3 was scored independently and blindly in three different area of each slide and then data were expressed as mean ± S.D, where ***p ≤ 0.001. iii Expression of galectin-3 in stage III breast cancer tissue specimens (receptor-matched non-smoker and smoker) as determined by western blot. Recombinant galectin-3 was used as a positive control to validate the molecular weight of the galectin-3 band in tissues. The number on top of blot represents arbitrary unit showing quantification of the galectin-3 expression as measured by ImageJ software. b Expression of gal3 in nicotine (1–100 μM) treated MCF-7 cells (48 h) as measured by quantitative RT-PCR. Relative mRNA expression was calculated according to the comparative ΔΔC(t) method. c Validation of gal3 expression in untreated and nicotine-treated (100 μM, 48 h) MCF7 cells by Western blot. The number on top of blot represents arbitrary unit showing quantification of the gal3 expression as measured by ImageJ software
α9nAChR directly modulates expression of galectin-3
As nicotine is known to induce the expression of α7nAChR in most cancers and α9nAChR in breast cancer, we quantified the levels of these two receptors using qRT-PCR. Nicotine treatment (100 μM, 48 h) induced only α9nAChR, but not α7nAChR (Fig. 3a). The induction of α9nAChR in the nicotine-treated cells was further confirmed by a Western blot analysis (Fig. 3b). To investigate if the tumors from smokers expressed higher amount of α9nAChR, tumor tissues from smoker and non-smoker groups were compared after immunostaining with an anti-α9nAChR antibody. α9nAChR positive cells were significantly (p < 0.001) higher in smokers than in non-smokers (Fig. 3c). Next, we investigated if α9nAChR was responsible for the nicotine-induced expression of galectin-3. Treatment of cells with nicotine in presence of a α9nAChR-specific siRNA did not result in an elevation of galectin-3 expression (Fig. 3d). Thus, α9nAChR is required for nicotine-induced galectin-3 expression.

Nicotine-induced α9nAChR expression. a Quantitative mRNA expression of α7nAChR and α9nAChR in nicotine-treated (100 μM for 48 h) MCF-7 cells by RT-PCR Relative mRNA expression was calculated according to the comparative ΔΔC(t) method. b Western blot showing protein expression of α9nAChR in nicotine-treated (100 μM for 48 h) MCF-7 cells. The number on top of blot represents arbitrary unit showing quantification of the α9nAChR expression as measured by ImageJ software. c Representative diagrams showing expression of α9nAChR in stage III tissues of breast cancer patients who never smoked (3 patients as described in Table 1) and smoked (5 patients) as determined by immunostaining with anti-α9nAChR antibodies. Adjacent bar graph shows quantitative expression of α9nAChR in these samples. Each sample was performed in duplicate or in triplicate. The number of cells showing positive staining of α9nAChR was scored independently and blindly in 3 different area of each slide and then data were expressed as mean ± S.D, where ***p ≤ 0.001. d Effect of α9nAChR silencing on the expression of galectin-3. Cells were treated with siRNA specific for α9nAChR or negative control siRNA and the cell extract was subjected to western blot for expression of galectin-3. The number on top of blot represents arbitrary unit showing quantification of the galectin-3 expression as measured by ImageJ software
Galectin-3 suppresses apoptosis by blocking mitochondrial damage
As galectin-3 is known to exert anti-apoptotic response in cancer cells, we investigated the effect of nicotine and galectin-3 on cell death. MCF-7 cells were treated with 1 μM STS following stimulation with nicotine (100 μM) in presence or absence of galectin-3 siRNA as described in the “Methods” section. Similar experiment was also performed with gal3 shRNA transfected MCF-7 cells. The efficacy of either gal3 siRNA or shRNA to knockdown gal3 expression was investigated by RT-PCR or Western blot and expression of gal3 was found down-regulated with treatment of either gal3 siRNA (Fig. 4a) or gal3 shRNA (Fig. 4b). Nicotine blocked STS-induced cell death by almost 50 % (p < 0.001), however, nicotine-treated cells regained their sensitivity to STS upon knocking down galectin-3 by either siRNA or shRNA (p < 0.001) (Fig. 4c).

Galectin-3 reduces apoptosis through promotion of mitochondrial stability. a Quantitative mRNA expression of galectin-3 in gene-specific or negative control siRNA treated MCF7 cells by RT-PCR. Relative mRNA expression based on GAPDH expression was calculated according to the comparative ΔΔC(t) method. b Western blot showing galectin-3 expression in galectin-3 specific si RNA treated MCF-7 cells. c MCF7 cells were challenged with known apoptotic inducer staurosporine (1 μM) for 48 h and then role of nicotine (100 μM) and galectin-3 was examined through viability study using WST1. Role of galectin-3 was investigated using galectin-3 specific siRNA (si-gal3) and shRNA (sh-gal3). d Amount of annexin-V positive cells was measured by flow cytometry as described in the “Methods” section, and data were presented as percent of annexin-V positive cells. e STS-induced DNA fragmentation of cells in the presence of nicotine alone or nicotine plus siRNA (galectin-3 specific or negative control) was measured on a colorimetric assay as described in the “Methods” section. F. The mitochondrial membrane potential was measured using JC1 dye and data were represented as the ratio of the emission 590 (red): 527 (green). All bar graphs showing quantitative data were represented as ±SD. ***p ≤ 0.001. NS not significant
The cytoprotective effect of nicotine and galectin-3 on drug-induced cell death was further corroborated through annexin V binding (Fig. 4d) and cytoplasmic histone-associated DNA fragmentation (Fig. 4e). In annexin V binding assay, STS alone induced apoptosis, which was reduced by almost fourfold when treated with nicotine plus STS (Fig. 4d). Consistent with the cell viability study, galectin-3 siRNA (si-gal3) abrogated the effect of nicotine on STS-induced apoptosis (Fig. 4d). Similar to the annexin V binding assay, nicotine reduced STS-induced DNA fragmentation of MCF-7 cells by almost 40 %, but galectin-3 siRNA or shRNA abrogated the effect of nicotine on STS-induced DNA fragmentation (Fig. 4e).
To investigate the effect of nicotine and galectin-3 on mitochondrial permeability transition, cells were treated with staurosporine (1 μM) alone or in the presence or absence of nicotine (100 μM) or in the presence of nicotine plus galectin-3 siRNA for 24 h and stained with JC-1 dye. JC-1 accumulates in mitochondria in potential-dependent manner, as indicated by a fluorescence emission shift from green (527 nm) to red (590 nm) under normal conditions. Mitochondrial depolarization is indicated by a decrease in the red/green fluorescent intensity ratio. The potential- sensitive color shift is due to concentration-dependent formation of red fluorescence J-aggregates [40]. STS-induced loss of mitochondrial membrane potential as indicated by the F590/F527 ratio was 0.77 compared to 1.3 for control cells (p < 0.001) (Fig. 4f). However, galectin-3 siRNA significantly (p < 0.001) abrogated the effect of nicotine on STS-induced loss of mitochondrial membrane potential (Fig. 4f). Taken together these results suggest a positive role for galectin-3 in promoting the integrity of mitochondrial membrane potential.
Nicotine induces side population in MCF-7 cells
Side-population (SP) cells within cancer cells represent a small fraction and are known to enrich cancer stem-like cells [41–43]. In these cells, certain ATP-binding cassette (ABC) transporters, including ABCG2/BCRP, ABCB1/MDR1, and ABCA3 can pump out or limit the intake of the fluorescent dyes such as Hoechst 33342 or DyeCycle Violet [41, 44]; whereas the rest of the cancer cells retain the dye. Thus, SP cells are analyzed by flow cytometer equipped with violet laser diode (VLD). DyeCycle Violet fluorescence is measured through blue and red band pass filter. DyeCycle Violet blue and red fluorescence are plotted allowing visualization of low fluorescence “tail” of cells (called “Side Population” cells) relative to the much larger bulk of DyeCycle Violet stained cells. SP of breast cancer cells has recently been shown to harbor stem cell-like properties with resistance to apoptosis [34–36]. Consistent with this finding, the MCF-7 cell line was found to contain SP cells (5.9 %), which showed as a distinct “tail” in the flow cytometry scatter plot (Fig. 5a). As inhibitors of ABC transporter are known to block SP population, we studied the effect of Verapamil on SP. As shown in Fig. 5a, Verapamil (50 μM) decreased the SP to 1.5 %. Nicotine-treated MCF-7 cells increased SP by 5.4-fold compared to untreated control cells (Fig. 5a, b).

Nicotine-induced side population (SP) in MCF7 cells and modulation of SP by galectin-3 and TWIST1. a Cells were treated with verapamil (50 μM), nicotine (100 μM), nicotine plus siRNA targeted galectin-3 or nicotine plus negative control siRNA, and stained with DyeCycle Violet and SP was measured using Dye cycle violet dye (DCV) on a flow cytometer. The SP gated populations are displayed as scatter plots for red (650 nm LP) vs. blue (450/40 nm BP) emissions of DCV (excited with 405 nm laser source). The ratio of the SP cells to total population of MCF-7 cells is indicated as percentage on each scatter plot (as represented by pink square). b Bar diagram showing results of side population cells in untreated and treated MCF-7 cells as described in a. Data were expressed as mean ± S.D, where ***p ≤ 0.001; **p ≤ 0.01, respectively. c Gene expression microarray showing i Heat map and ii The Scatter Plot. i The heat map is depicting the overall impression of the change in gene regulation of PCR array specific genes. The color code specifies the magnitude of change of gene expression (Green to Red for upregulation). In the heat map, left panel is showing the basal gene expression of the untreated MCF-7 cells and right panel represents nicotine-treated group. ii MCF-7 cells were treated with nicotine (100 μM, 48 h) and expression of all the genes (genes included in PCR array 96 well plate) was measured in comparison to the untreated control. The scatter plot shows the expression level (2−ΔΔCt) of each gene in the nicotine-treated cells compared to the untreated control. The black line indicates fold changes (2−ΔΔCt) of 1. TWIST1 was indicated as most up-regulated gene. The expression fold of each gene was shown in Table 2. d Validation of TWIST1 expression in nicotine-treated cells by Western blot. The number on top of blot represents arbitrary unit showing quantification of the TWIST1 expression as measured by ImageJ software. e Expression of TWIST1. Representative diagrams showing expression of TWIST1 in tissues of stage III breast cancer patients who never smoked (3 patients as described in Table 1) and smoked (5 patients) as determined by immunostaining with anti-TWIST1 antibody. Adjacent bar graph shows quantitative expression of TWIST1 in these samples. Each sample was performed in duplicate or in triplicate. The number of cells showing positive staining of TWIST1 was scored independently and blindly in three different area of each slide and then data were expressed as mean ± S.D, where ***p ≤ 0.001. f Effect of TWIST1 silencing on SP. Cells were treated with nicotine or nicotine plus si-TWIST1 and SP was measured as described in a
Effect of galectin-3 and TWIST1 on nicotine-induced side population in MCF-7 cells
To investigate the effect of galectin-3 on SP, MCF-7 cells were treated with nicotine plus galectin-3 siRNA (si-gal3). The si-gal3 abrogated the effect of nicotine on SP by 2.4-fold suggesting the importance of galectin-3 on induction of SP (Fig. 5a, b). To investigate if any other genes could be involved in inducing SP, RNA from nicotine-treated MCF-7 cells was screened for the expression for a few genes involved in cancer pathways including epithelial-to-mesenchymal transition using a microarray. Notably, expression of TWIST1 was remarkably high (15-fold) (Fig. 5c; see also Table 2). This was corroborated by Western blot analysis as nicotine-treated cells showed higher amount of TWIST1 protein compared to the untreated cells (Fig. 5d). Higher numbers of TWIST1 positive cells were present in the breast tumors of smokers compared to non-smokers (Fig. 5e).
To investigate the effect of TWIST1 on SP, MCF-7 cells were treated with nicotine plus siRNA targeted to TWIST1. As shown in Fig. 5f, siRNA of TWIST1 reduced nicotine-induced SP by ~2.8-fold % (p < 0.001) suggesting the importance of TWIST1 for the existence of SP cells.
STAT3 modulates expression of galectin-3 and TWIST1
Since STAT3 is an important regulator of TWIST1 [45], we investigated if nicotine treatment could induce STAT3 activation. In nicotine-treated cells, an increased pY705-STAT3 level was observed, with no change in the total STAT3-levels (Fig. 6a). Consistent with this observation, higher numbers of pY705-STAT3 positive cells were found in the breast tumors derived from smokers than matching non-smokers (Fig. 6b). We next tested the relevance of STAT3 to TWIST1 expression by RNAi-mediated silencing of STAT3. siRNA of STAT3 was first validated as the shRNA treated cells showed significant drop in STAT3 expression (Fig. 6c). As shown in Fig. 6d, sh1-STAT3 led to a decline in the nicotine-induced expression of TWIST1. To investigate if galectin-3 expression was modulated by TWIST1 and STAT3, galectin-3 expression was determined in cells depleted of these factors. Knockdown of TWIST1 did not affect galectin-3 expression (data not shown). However, cells depleted of STAT3 (using 3 shRNAs) strongly suppressed the nicotine-induced expression of galectin-3 (Fig. 6e) suggesting a requirement for STAT3. Transcription factor binding site analyses of the LGALS3 promoter identified a potential STAT3 binding sequence (TTTCTGGGAA) from −2734 to −2725 bp (Fig. 6f). A ChIP assay was then performed using STAT3 antibody to determine if STAT3 bound to the LGALS3 promoter in a nicotine-dependent manner. As shown in Fig. 6g, LGALS3 promoter was detected in STAT3-ChIP product. The positive control promoter, TWIST1, was also detected in ChIP analyses (Fig. 6g), since it is known to be a transcriptional target of STAT3 [45]. Neither LGALS3 nor TWIST1 was detected in the negative control (CHIP assay without STAT3 antibody) (data not shown).

Nicotine-induced STAT3 activation and promoted expression of galectin-3 and TWIST1. a Western blot showing STAT3 activation as identified by positive staining with anti-phosphorylated (at Tyr705) STAT3 antibody. The number on top of blot represents arbitrary unit showing quantification of STAT3p expression as measured by ImageJ software. b i Representative diagrams showing expression of STAT3p in tissues of stage III breast cancer patients who never smoked (3 patients as described in Table 1) and smoked (5 patients) as determined by immunostaining with anti-phosphorylated (at Tyr705) STAT3 antibody. Adjacent bar graph shows quantitative expression of STAT3p in these samples. Each sample was performed in duplicate or in triplicate. The number of cells showing positive staining of STAT3p was scored independently and blindly in 3 different area of each slide and then data were expressed as mean ± S.D, where ***p ≤ 0.001. c Quantitative mRNA expression of STAT3 in gene-specific or negative control siRNA treated MCF7 cells by RT-PCR. Relative mRNA expression based on GAPDH expression was calculated according to the comparative ΔΔC(t) method. d, e Effect of STAT3 silencing on expression of TWIST1 (d) and galectin-3 (e). Cells were treated with nicotine or nicotine plus shRNA targeted to STAT3 and each extract was subjected to Western blot detection of Twist and galectin-3. The number on top of blot represents arbitrary unit showing quantification of TWIST1 (d) and galectin-3 (e) expression as measured by ImageJ software. f Partial sequence of the galectin-3 promoter comprising −2756 bp to −2688 bp upstream of the transcription site. Sequence in red color represents STAT3 binding motif. g Chromatin immunoprecipitation (ChIP) with anti-phosphorylated (Tyr705) STAT3 antibody and the precipitated DNA was subjected to PCR with primers located at promoters of TWIST1 and galectin-3 (LGALS3)
STAT3 modulates nicotine-induced SP and anti-apoptotic property in MCF-7 cells
Since STAT3 activation is required for galectin-3 expression, we investigated if silencing of STAT3 could influence on SP and apoptosis. Cells were treated with nicotine alone (100 μM, 48 h) or in the presence of sh1-STAT3 and analyzed the SP. As expected, nicotine-induced SP was significantly (p < 0.001) reduced in presence of sh1-STAT3 (Fig. 7a). We next analyzed apoptotic sensitivity of cells upon STS treatment in the presence of nicotine. As shown in Fig. 7b, nicotine reduced STS-induced cell death by ~50 %, but silencing of STAT3 abrogated the effect of nicotine completely and promoted (72 %) cell death.

STAT3 promotes nicotine-induced side population (SP) and cell viability. a MCF-7 cells were treated with nicotine (100 μM, 48 h) or nicotine plus sh1-STAT3 and SP was analyzed using Dye cycle violet dye (DCV). b Cell viability of the treated cells was studied using WST-1. In both cases, data were represented as ± SD, ***p ≤ 0.001. Staurosporine was used as an apoptosis inducer
α9nAChR directly modulates STAT3 activation
To investigate if α9nAChR modulate STAT3 activation, α9nAChR was silenced in MCF7 cells using RNAi and STAT3 phosphorylation was analyzed. Knockdown of α9nAChR prevented nicotine-induced STAT3 activation (Fig. 8a). We also studied the effect of α9nAChR silencing on cell viability and SP formation. As shown in Fig. 8b, nicotine prevented STS-induced cell death from 39 to 17 %. Silencing of α9nAChR abrogated the effect of nicotine completely and promoted cell death (48 %). Flow cytometry revealed that knockdown of α9nAChR reduced SP by 2.5-fold (p < 0.001) compared nicotine alone (Fig. 8c).

α9nAChR promotes nicotine-induced STAT3 activation, cell viability, side population (SP), and formation of mammospheres. a MCF-7 cells were treated with nicotine (100 μM, 48 h) or nicotine plus si-α9nAChR and STAT3 activation was measured by anti-phosphorylated (at Tyr705) STAT3 antibody on Western blot. The number on top of blot represents arbitrary unit showing quantification of the STAT3p expression as measured by ImageJ software. b Cell viability of the treated cells was studied using WST-1. The data was represented as ±SD, ***p ≤ 0.001. Staurosorine used as an apoptosis inducer. c Effect of α9nAChR silencing on SP. Cells were treated with nicotine (100 μM, 48 h) or nicotine plus si-α9nAChR and SP was measured using Dye cycle violet dye (DCV). d, e Effect of nicotine and silencing of various genes on mammosphere formation. MCF-7 cells were treated with siRNA targeted to each gene for 48 h. After washing, equal number of cells from each treatment was incubated in Mammocult medium for 48 h and spheres were counted. In d, a representative mammosphere that obtained from the nicotine-treated MCF-7 cells is shown
As SP cells are known to possess cancer stem-like property and are resistant to apoptosis [30–32], we investigated mammosphere formation in untreated and nicotine-treated MCF-7 cells in absence or in the presence of various siRNAs. Nicotine-treated MCF-7 cells readily formed several mammospheres (Fig. 8d), but si-RNAs specific for galectin-3, α9nAChR, TWIST1, and STAT3 strongly reduced formation of mammospheres (80–94 % reduction, p < 0.001) (Fig. 8e).
Discussion
Galectin-3 regulates multiple biological functions, such as cell proliferation, tumor progression and metastasis, and apoptosis [13–16, 23], but the detailed mechanisms of its effects and the corresponding signaling molecules are not well understood. Increased expression of galectin-3 in many cancers and its role in promotion of tumor growth and metastasis [17–22] suggests that galectin-3 may serve as an attractive target for cancer therapy. In fact, a few studies on galectin-3 targeted therapy were performed to document therapeutic effects of synthetic carbohydrate-based small molecule inhibitors of galectin-3 [26–29]. Although the expression of galectin-3 in early stages of prostate cancer is slightly down-regulated by methylation of its promoter DNA [31, 46], it promotes prostate cancer growth and metastasis possibly by modulating host immune system [16]. In this study, we show that expression of galectin-3 in breast cancer cells such as MCF-7 is modulated by nicotine or its receptor, α9nAChR. Nicotine-induced up-regulation of galectin-3 might be involved in the promotion of stem cell property and apoptosis resistance.
Nicotine interacts with nAChR and functions in muscle or at the nervous system to elicit tobacco addiction [47, 48]. Among many isoforms of nAChR, α7nAChR has been linked to progression of many cancers [2]. However, recent reports suggest that α9nAChR is associated with the progression of breast cancer [6, 49]. Consistent with this report, we observed nicotine-induced up-regulation of α9nAChR, but not α7nAChR, in MCF-7 cells. Up-regulation of α9nAChR was also observed in breast cancers from patients, who were smokers compared to those from non-smokers. Nicotine-dependent activation of MAPK and PI3K/Akt were reported as two important signaling pathways downstream to α9nAChR [49, 50]. These signaling pathways activate estrogen receptor, which then interacts with two other transcription factors AP1 and VDR and bind to the promoter of α9nAChR forming a strong positive feedback loop. In this study, we present α9nAChR-mediated STAT3 activation as a new signaling cascade in breast cancer cells exposed to nicotine. Microarray analyses of nicotine-treated MCF-7 cells for a few genes involved in cancer pathways, revealed a remarkable up-regulation of TWIST1. As STAT3 is an important transcription regulator of TWIST1, we investigated if nicotine treatment could cause STAT3 activation. Indeed, STAT3 was activated through α9nAChR in MCF-7 cells exposed to nicotine. However, nicotine-induced STAT3 activation through α7nAChR is known in other cancers such as pancreatic [4] and bladder [51] cancers. In pancreatic cancer, cigarette-smoke or nicotine causes up-regulation of MUC4 mucin via α7nAChR/JAK2/STAT3 pathway and promotes metastasis [4].
In the cytokine signaling pathways, phosphorylation of STAT3 at Tyr705 promotes its dimerization and translocation from the cytoplasm to nucleus [52]. In the nucleus, STAT3 binds to target genes and induces the transcription of several genes associated with cell proliferation and anti-apoptosis [52, 53]. We have been working on the gene regulation and functional characterization of galectin-3 over the years [16, 31, 46, 54–56]. Several studies suggest that galectin-3 is involved in tumor cell adhesion, proliferation, angiogenesis, cancer progression, and metastasis [13, 16–20]. Moreover, intracellular galectin-3 suppresses drug induced apoptosis and anoikis [23, 57]. However, it is unknown if galectin-3 is regulated by α9nAChR or STAT3 in nicotine-induced cancer cells. Here, we showed that nicotine-induced α9nAChR activates STAT3 which induces the transcription of LGALS3 in MCF-7 breast cancer cells. We also found that galectin-3 prevents mitochondrial damage under conditions of chemotherapeutic challenge.
Recent studies suggest that side-population (SP) cells within cancer cells are enriched with cancer stem-like properties and are resistant to apoptosis [34–36]. Nicotine treatment of MCF-7 cells has recently been shown to increase cancer stem cell [58], although the mechanisms are unclear. In our study, galectin-3 was found critical for the enrichment of nicotine-induced SP of MCF-7 cells. Moreover, expression of TWIST1 modulated by STAT3 enriched SP in MCF-7 cells. Drug- or chemo-resistance is also positively correlated with nicotine or cigarette smoke in many cancers [5, 10, 59–61]. For example, nicotine-induced chemo-resistance in non-small cell lung cancer (NSCLC) occurs through up-regulation of XIAP and surviving [5]. In lung cancer, nicotine induces chemo-resistance by modulating mitochondrial signaling [59]. In bladder cancer, nicotine-induced chemo-resistance is mediated by activation of STAT3 and down regulation of ERK1/2 via anchor and beta-adrenoceptors [10]. In this study, we present evidence that nicotine treatment induces expression of α9nAChR followed by activation of STAT3. Activated STAT3 directly binds to LGALS3 and TWIST1 promoters and induces transcriptional up-regulation of these genes resulting to delayed apoptosis and enriched SP of breast cancer cells.
Taken all together, α9nAChR acts as an upstream signaling molecule that modulates expression of galectin-3 through activation of STAT3 and manifest the functional significance of nicotine in progression of breast cancer (Fig. 9). Thus, galectin-3 or its upstream signaling molecule STAT3 or α9nAChR could be potential target in suppressing nicotine-induced chemo-resistance in breast cancer.

A schematic diagram showing nicotine-induced signaling in MCF-7 cells. Nicotine treatment induces expression of α9nAChR and activates STAT3 through phosphorylation at Tyr705. Activated STAT3 migrates to nucleus and binds to the promoter of galectin-3 and TWIST1 resulting into transcriptional up-regulation of these genes
Abbreviations
- α9nAChR:
-
α9 isoform of nicotinic acetylcholine receptor
- LGALS3:
-
Galectin-3
- gal3:
-
galectin-3
- STS:
-
Staurosporine
- SP:
-
Side population
References
Adhikari B, Kahende J, Malarcher A, Pechacek T, Tong V (2008) Smoking-attributable mortality, years of potential life lost, and productivity losses—United States, 2000–2004. Morbidity and Mortality Weekly Rep 2008, 57:1226–1228
Singh S, Pillai S, Chellappan S (2011) Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J Oncol 2011:456743
Nishioka T, Kim HS, Luo LY, Huang Y, Guo J, Chen CY (2011) Sensitization of epithelial growth factor receptors by nicotine exposure to promote breast cancer cell growth. Breast Cancer Res 13:R113
Momi N, Ponnusamy MP, Kaur S, Rachagani S, Kunigal SS, Chellappan S, Ouellette MM, Batra SK (2013) Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through α9 nAChR-mediated MUC4 upregulation. Oncogene 32:1384–1395
Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S (2006) Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and surviving. Proc Natl Acad Sci USA 103:6332–6337
Shih YL, Liu HC, Chen CS, Hsu CH, Pan MH, Chang HW, Chen FC, Ho CT, Yang YY, Ho YS (2010) Combination treatment with luteolin and quercetin enhances antiproliferative effects in nicotine-treated MDA-MB-231 cells by down regulating nicotinic acetylcholine receptors. J Agric Food Chem 58:235–241
Levy DE, Darnell JE Jr (2002) Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662
Takemoto S, Ushijima K, Kawano K, Yamaguchi T, Terada A, Fujiyoshi N, Nishio S, Tsuda N, Ijichi M, Kakuma T et al (2009) Expression of activated signal transducer and activator of transcription-3 predicts poor prognosis in cervical squamous-cell carcinoma. Br J Cancer 101:967–972
Ryu K, Choy E, Yang C, Susa M, Hornicek FJ, Mankin H, Duan Z (2010) Activation of signal transducer and activator of transcription 3 (Stat3) pathway in osteosarcoma cells and overexpression of phosphorylated-Stat3 correlates with poor prognosis. J Orthop Res 28:971–980
Chen RJ, Ho YS, Guo HR, Wang YJ (2010) Long-term nicotine exposure-induced chemoresistance is mediated by activation of Stat3 and downregulation of ERK1/2 via nAChR and beta-adrenoreceptors in human bladder cancer cells. Toxicol Sci 115:118–130
Macha MA, Matta A, Chauhan SS, Siu KW, Ralhan R (2011) Guggulsterone (GS) inhibits ST/Nicotine modulated NF-kB and STAT3 pathways in oral cancer SCC4 cells. Carcinogenesis 32:368–380
Xu TY, Guo LL, Wang P, Song J, Le YY, Viollet B, Miao CY (2012) Chronic exposure of nicotine enhances insulin sensitivity though α7 nicotinic acetylcholine receptor-STAT3 pathway. PLoS ONE 7:e51217
Liu FT, Rabinovich GA (2005) Galectins as modulators of tumor progression. Nat Rev Cancer 5:29–41
Radosavljevic G, Volarevic V, Jovanovic I, Milovanovic M, Pejnovic N, Arsenijevic N, Hsu DK, Lukic ML (2012) The roles of galectin-3 in autoimmunity and tumor progression. Immunol Res 52:100–110
Rabinovich GA, van Kooyk Y, Cobb BA (2012) Glycobiology of immune responses. Ann NY Acad Sci 1253:1–15
Guha P, Kaptan E, Bandypadhyaya G, Kaczanowska S, Davila E, Thompson K, Martin SS, Kalvakolanu DK, Vasta GR, Ahmed H (2013) Cod glycopeptides with picomolar affinity to galectin-3 suppresses T-cell apoptosis and prostate cancer metastasis. Proc Natl Acad Sci USA 110:5052–5057
Hsu DK, Dowling CA, Jeng KC, Chen JT, Yang RY, Liu FT (1999) Galectin-3 expression is induced cirrhotic liver and hepatocellular carcinoma. Int J Cancer 81:519–526
Miyazaki J, Hokari R, Kato S, Tsuzuki Y, Kawaguchi A, Nagao S, Itoh K, Miura S (2002) Increased expression of Galectin-3 in primary gastric cancer and the metastatic lymph nodes. Oncol Rep 9:1307–1312
Yoshimura A, Gemma A, Hosoya Y, Komaki E, Hosomi Y, Okano T, Takenaka K, Matuda K, Seike M, Uematsu K et al (2003) Increased expression the LGALS3 (galectin 3) gene in human non-small-cell lung cancer. Genes Chromosomes Cancer 37:159–164
Saussez S, Decaestecker C, Mahillon V, Cludts S, Capouillez A, Chevalier D, Vet HK, André S, Toubeau G, Leroy X et al (2008) Galectin-3 up-regulation during tumor progression in head and neck cancer. Laryngoscope 118:1583–1590
Lotz MM, Andrews CW Jr, Korzeliu CA, Lee EC, Steele GD Jr, Clarke A, Mercurio AM (1993) Decreased expression of Mac-2 (carbohydrate binding protein 35) and loss of its localization are associated with the neoplastic progression of colon carcinoma. Proc Natl Acad Sci USA 90:3466–3470
Honjo Y, Inohara H, Akahani S, Yoshii T, Takenaka Y, Yoshida J, Hattori K, Tomiyama Y, Raz A, Kubo T (2000) Expression of cytoplasmic galectin-3 as a prognostic marker in tongue carcinoma. Clin Cancer Res 6:4635–4640
Nakahara S, Oka N, Raz A (2005) On the role of galectin-3 in cancer apoptosis. Apoptosis 10:267–275
Wang Y, Nangia-Makker P, Tait L, Balan V, Hogan V, Pienta KJ, Raz A (2009) Regulation of prostate cancer progression by galectin-3. Am J Pathol 174:1515–1523
Burlacu A (2003) Regulation of apoptosis by by Bcl-2 family proteins. J Cell Mol Med 7:249–257
Glinsky GV, Price JE, Glinsky VV, Mossine VV, Kiriakova G, Metcalf JB (1996) Inhibition of human breast cancer metastasis in nude mice by synthetic glycoamines. Cancer Res 56:5319–5324
Johnson KD, Glinskii OV, Mossine VV, Turk JR, Mawhinney TP, Anthony DC, Henry CJ, Huxley VH, Glinsky GV, Pienta KJ, Raz A, Glinsky VV (2007) Galectin-3 as a potential therapeutic target in tumors arising from malignant endothelia. Neoplasia 9:662–670
Glinsky VV, Kiriakova G, Glinskii OV, Mossine VV, Mawhinney TP, Turk JR, Glinskii AB, Huxley VH, Price JE, Glinsky GV (2009) Synthetic galectin-3 inhibitor increases metastatic cancer cell sensitivity to taxol-induced apoptosis in vitro and in vivo. Neoplasia 11:901–909
Glinskii OV, Sud S, Mossine VV, Mawhinney TP, Anthony DC, Glinsky GV, Pienta KJ, Glinsky VV (2012) Inhibition of Prostate Cancer Bone Metastasis by Synthetic TF Antigen Mimic/Galectin-3 Inhibitor Lactulose-l-Leucine. Neoplasia 14:65–73
Sun P, Nallar SC, Kalakonda S, Lindner DJ, Martin SS, Kalvakolanu DV (2009) GRIM-19 inhibits v-Src-induced cell motility by interfering with cytoskeletal restructuring. Oncogene 28:1339–1347
Ahmed H, Cappello F, Rodolico V, Vasta GR (2009) Evidence of heavy methylation in the galectin-3 promoter in early stages of prostate-adenocarcinoma: development and validation of a methylated marker for early diagnosis of prostate cancer. Transl Oncol 2:146–156
Banerjee A, Lang JY, Hung MC, Sengupta K, Banerjee SK, Baksi K, Banerjee DK (2011) Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J Biol Chem 286:29127–29138
Sierra J, Villagra A, Paredes R, Cruzat F, Gutierrez S, Javed A, Arriagada G, Olate J, Imschenetzky M, Van Wijnen AJ et al (2003) Regulation of bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. Mol Cell Biol 23:3339–3351
Guo J, Ibaragi S, Zhu T, Luo LY, Hu GF, Huppi PS, Chen CY (2008) Nicotine promotes mammary tumor migration via a signaling cascade involving PKC and ccd42. Cancer Res 68:8473–8481
Kondo T, Setoguchi T, Taga T (2004) Persistence of a small subpopulation of cancer-stem like cells in the C6 glioma cell line. Proc Natl Acad Sci USA 101:781–786
Telford WG, Bradford J, Godfrey W, Robey RW, Bates SE (2007) Side population analysis using a violet-excited cell-permeable DNA binding dye. Stem Cells 25:1029–1036
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 17:1253–1270
Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D et al (2009) Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell line. Int J Cancer 124:36–45
Hung CS, Peng YJ, Wei PL, Lee CH, Su HY, Ho YS, Lin SY, Wu CH, Chang YJ (2011) The alpha9 nicotinic acetylcholine receptor is the key mediator in nicotine-enhanced cancer metastasis in breast cancer cells. J Exp Clin Med 3:283–292
Han J, Goldstein LA, Gastman BR, Rabinowich H (2006) Interrelated roles for Mcl-1and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem 281:10153–101163
Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E III, Zhang Y (2007) Activation of the PTEN/Mtor/STAT3 pathway in breast cancer stem-like cells in required for viability and maintenance. Proc Natl Acad Sci USA 104:16158–16163
Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG (2005) Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic. Cancer Res 65:6207–6219
Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK (2004) A distinct “side population” of cells with druf efflux capacity in human tumor cells. Proc Natl Acad Sci USA 101:14228–14233
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H et al (2001) The ABC transporter Bcrp1/ABCG2 is expressed in wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7:1028–1034
George ZC, WeiZhou Z, Mei Sun QW, Domenico C, Mena M, LiMei X, Carliann C, Jin QC, Lu-Hai W (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 283:14665–14673
Ahmed H, Banerjee PP, Vasta GR (2007) Differential expression of galectins in normal, benign and malignant prostate epithelial cells: silencing of galectin-3 expression in prostate cancer by its promoter methylation. Biochem Biophys Res Commun 358:241–246
Alkondon M, Albuquerque EX (2004) The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog Brain Res 145:109–120
Arredondo J, Chernyavsky AI, Grando SA (2006) Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamine on immortalized oral epithelial cells. Cancer Biol Ther 5:511–517
Wu CH, Lee CH, Ho YS (2011) Nicotinic acetylcholine receptor-based blockade: application of molecular targets for cancer therapy. Clin Cancer Res 17:3533–3541
Lee CH, Chang YC, Chen CS, Tu SH, Wang YJ, Chen LC, Chang YJ, Wei PL, Chang HW, Chang CH et al (2011) Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces α9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res Treat 129:331–345
Chen RJ, Ho YS, Guo HR, Wang YJ (2008) Rapid activation of Stat3 and ERK1/2 by nicotine modulates cell proliferation in human bladder cancer cells. Toxicol Sci 104:283–293
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19:2474–2488
Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL (2002) Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21:7611–7618
Mercer N, Ahmed H, Etcheverry SB, Vasta GR, Cortizo AM (2007) Regulation of advanced glycation end product (AGE) receptors and apoptosis by AGEs in osteoblast-like cells. Mol Cell Biochem 306:87–94
Ahmed H (2010) Promoter methylation in prostate cancer and its application for the early detection of prostate cancer using serum and urine samples. Biomark Cancer 2:17–33
Ahmed H, Guha P, Kaptan E, Bandyopadhyaya G (2011) Galectin-3: a potential target for cancer prevention. Trends Carbohydr Res 3:13–22
Fukumori T, Oka N, Takenaka Y, Nangia-Makker P, Elsamman E, Kasai T, Shono M, Kanayama HO, Ellerhors J, Lotan R et al (2006) Galectin-3 regulates mitochondrial stability and antiapoptotic function in response to anticancer drug in prostate cancer. Cancer Res 66:3114–3119
Hirata N, Sekino Y, Kanda Y (2010) Nicotine increases cancer stem cell population in MCF-7 cell. Biochem Biophys Res Commun 403:138–143
Zhang J, Kamdar O, Le W, Rosen GD, Upadhyay D (2009) Nicotine induces resistance to chemotherapy by modulating mitochondrial signaling in lung cancer. Am J Respir Cell Mol Biol 40:135–146
Shen T, Le W, Yee A, Kamdar O, Hwang PH, Upadhyay D (2010) Nicotine induces resistance to chemotherapy in nasal epithelial cancer. Am J Rhinol Allergy 24:73–77
An Y, Kiang A, Lopez JP, Kuo SZ, Yu MA, Abhold EL, Chen JS, Wang-Rodriguez J, Ongkeko WM (2012) Cigarette smoke promotes drug resistance and expansion of cancer stem cell-like side population. PLoS ONE 7:e47919
Acknowledgments
We are grateful to Dr. Aditi Banerjee for her help on the scratch-wound healing assay. This study was supported by the University of Maryland Start-up fund and in part by the NIH Grants RO3CA133935 and R41CA141970 to H.A; and R01CA105005 and Cigarette Restitution Funds from the University of Maryland Greenebaum Cancer Center to D.V.K.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Prasun Guha and Gargi Bandyopadhyaya have contributed equally.
Rights and permissions
About this article
Cite this article
Guha, P., Bandyopadhyaya, G., Polumuri, S.K. et al. Nicotine promotes apoptosis resistance of breast cancer cells and enrichment of side population cells with cancer stem cell-like properties via a signaling cascade involving galectin-3, α9 nicotinic acetylcholine receptor and STAT3. Breast Cancer Res Treat 145, 5–22 (2014). https://doi.org/10.1007/s10549-014-2912-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-014-2912-z