
Abstract
The clinical importance of CYP2D6 genotype as predictor of tamoxifen efficacy is still unclear. Recent genotyping studies on CYP2D6 using DNA derived from tumor blocks have been criticized because loss of heterozygosity (LOH) in tumors may lead to false genotype assignment. Postmenopausal early breast cancer patients who were randomized to receive tamoxifen, followed by exemestane in a large randomized controlled trial were genotyped for five CYP2D6 alleles. CYP2D6 genotypes and phenotypes were related to disease-free survival during tamoxifen use (DFS-t) in 731 patients. By analyzing microsatellites flanking the CYP2D6 gene, patients whose genotyping results were potentially affected by LOH were excluded. In addition, exploratory analyses on 24 genetic variants of other metabolic enzymes and the estrogen receptor were performed. For the CYP2D6 analysis, only 2.3 % of the samples were excluded, because influence of LOH could not be ruled out. No association was found between the CYP2D6 genotype or predicted phenotype and DFS-t (poor vs. extensive metabolizers: unadjusted hazard ratio 1.33, 95 % CI 0.52–3.43; P = 0.55). DFS-t was associated with UGT2B15*2 (Vt/Vt + Wt/Vt vs. Wt/Wt: adjusted hazard ratio 0.47, 95 % CI 0.25–0.89; P = 0.019) and the estrogen receptor-1 polymorphism ESR1 PvuII (gene–dose effect: adjusted hazard ratio 1.63, 95 % CI 1.04–2.54; P = 0.033). In postmenopausal early breast cancer patients treated with adjuvant tamoxifen followed by exemestane neither CYP2D6 genotype nor phenotype did affect DFS-t. This is in accordance with two recent studies in the BIG1-98 and ATAC trials. Our study is the first CYP2D6 association study using DNA from paraffin-embedded tumor tissue in which potentially false interpretation of genotyping results because of LOH was excluded. Polymorphisms in the estrogen receptor-1 and UGT2B15 may be associated with tamoxifen efficacy, but these findings need replication.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
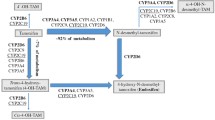
Adjuvant treatment with tamoxifen effectively decreases breast cancer recurrence. Responsiveness to tamoxifen may be partially based on a patient’s ability to metabolize tamoxifen to its active metabolites 4-OH tamoxifen and the more abundant endoxifen. Biotransformation to endoxifen is mainly mediated by cytochrome P450 2D6 (CYP2D6), but several other enzymes are also involved in the formation of both active metabolites [1]. Supported by in vitro experiments, several cohort studies raised expectations that CYP2D6 genotype would become a clinically useful predictive marker for tamoxifen therapy [2–7]. Still, clinical data on both the influence of the CYP2D6 genotype and CYP2D6 inhibitors on tamoxifen response are conflicting and some important issues remain unresolved [8–17]. First, endoxifen’s activity in vivo is uncertain, while its activity in vitro is apparent [10]. Second, if endoxifen is the effective component in humans, the critical concentration needed for activity is uncertain, although a threshold of 5.9 ng/ml has recently been suggested [18]. Finally, large inter-individual variance in endoxifen plasma concentration is only partially explained by CYP2D6 activity [3, 7, 19]. While the tamoxifen metabolism is complex, variants of genes encoding other metabolic enzymes may also influence blood levels of 4-OH tamoxifen and/or endoxifen (Fig. 1) [1, 20, 21]. Although it has been suggested that publications showing no association between the CYP2D6 genotype and tamoxifen efficacy are confounded, positive studies often also lack the ability to adjust for possible confounders [10]. The recently performed large clinical trials that have compared the efficacy of tamoxifen with an aromatase inhibitor contain conscientiously collected patient data and are therefore well suited for pharmacogenetic analyses (e.g., ATAC, BIG1-98, IES, and TEAM). Recently, two studies on patient cohorts of the BIG1-98 and ATAC trials did not result in an association between CYP2D6 genotype and tamoxifen efficacy [16, 17]. However, the CYP2D6 genotype analysis within the BIG1-98 patient cohort has been heavily criticized for using DNA from tumor instead of normal tissue [22–25]. In the BIG1-98 study, the apparent deviation from the Hardy–Weinberg equilibrium may be explained by the loss of heterozygosity (LOH) of the CYP2D6 locus on chromosome 22q13 in tumor tissue [22, 26]. To justify the use of DNA retrieved from FFPE tumor blocks instead of DNA from normal tissue, a false homozygous genotype call because of LOH of chromosome 22q13 should be ruled out when CYP2D6 germline genetic variations are investigated.

Tamoxifen metabolism. CYP cytochrome P450 isoenzyme, SULT sulfotransferase, UGT UDP-glucuronosyltransferase, NR1 nuclear receptor subfamily 1, PXR pregnane X receptor, CAR constitutive androstane receptor
In the Tamoxifen Exemestane Adjuvant Multinational (TEAM) trial, the efficacy of tamoxifen followed by exemestane (after 2.5–3 years) was compared to that of 5 years of exemestane in postmenopausal hormone receptor positive early breast cancer patients [27]. In the current study, we investigated possible associations between the CYP2D6 genotype/phenotype and disease-free survival in a Dutch cohort of the TEAM trial. Patients were excluded from our analysis when the possibility of a false CYP2D6 genotype because of LOH in the tumor could not be ruled out. In addition, associations with other genetic variants of metabolic enzymes and the estrogen receptor were explored.
Methods
Patients
Of in total 9,779 postmenopausal early stage breast cancer patients enrolled in the TEAM trial, 2,753 were included in The Netherlands and 1,379 were randomized to tamoxifen (20 mg once daily) with a planned switch to exemestane after 2.5–3 years. Tumor blocks of 746 of these 1,379 patients (54.1 %) were available for genotyping. Information on tumor and patient characteristics including concomitant medication use, the tamoxifen start date as well as planned and unplanned stop dates were locally registered on case record forms and centrally collected at the datacenter in Leiden, The Netherlands. The current pharmacogenetic study was separately approved by the central medical ethics review board of the Erasmus University Medical Center in Rotterdam, The Netherlands.
Endpoint
The endpoint of the core TEAM trial was disease-free survival (DFS-c), defined as the time from randomization to locoregional or distant recurrence, second breast cancer, or death without recurrence [27]. To avoid effect modification by subsequent aromatase inhibitor use, a new endpoint was created for the purpose of this pharmacogenetic study, being disease-free survival defined as the time from the tamoxifen start date to the tamoxifen discontinuation date (DFS-t). Patients were censored at the time of tamoxifen discontinuation for reasons other than an event (e.g., planned or unplanned switch to exemestane or another aromatase inhibitor or intolerable side effects) or loss to follow-up. The original endpoint of complete disease-free survival (DFS-c), as used in the core TEAM trial analysis, was used in a sensitivity analysis [27].
Genotyping
Germline genetic variants in candidate genes of enzymes involved in the tamoxifen metabolism and of the estrogen receptor (Fig. 1) were selected based on assumed clinical relevance, high allelic frequency or the assumption that nonsynonymous amino acid change leads to altered protein functionality. The polymorphisms included in our analyses are listed in Table 1. Genotyping was performed on formalin fixed paraffin-embedded tumor (FFPE) tissue as described previously [28]. In brief, from three slides of 20 μm DNA was extracted with the Maxwell forensic DNA isolation kit (Promega, Leiden, The Netherlands). Before genotyping a pre-amplification step was used to increase the percentage of successfully genotyped samples without loss of reliability and with minimal use of DNA mass [28]. For genotyping Taqman assays (Applied Biosystems, Foster City, CA, USA) were used on the Biomark (Fluidigm, San Fransisco, USA). In case of failure of genotyping using the Taqman based method, pyrosequencing was performed (Qiagen, Chatsworth, CA, USA) on a Pyrosequencer 96 MA (Biotage, Uppsala, Sweden). CYP2D6 genotypes were translated to predicted phenotypes (extensive, intermediate, or poor metabolizer) as described in the supplementary methods section.
Loss of heterozygosity
The ratio between tumor and germline DNA in a sample derived from FFPE tumor tissue differs between samples. A high percentage of tumor DNA may result in falsely called genotypes because of LOH in the tumor. If a certain germline homozygous CYP2D6 genotype was assumed, while in fact one of the alleles has been lost in tumor but not in normal tissue a false test is the result [22]. To avoid such incorrect interpretation of CYP2D6 genotyping results, three microsatellite markers D22S276, D22S2284, and D22S423 near the CYP2D6 gene on chromosome 22q13 with a high frequency of heterozygosity (>80 %) were additionally determined (Table 4 supplementary files). The chance that a patient is homozygous for all three markers would be less than 0.203 = 0.8 %. Thus, in nearly all patients including those with a homozygous germline CYP2D6 genotype, heterozygosity should be demonstrated for ≥1 microsatellite markers. We hypothesized that LOH of the CYP2D6 gene would also lead to LOH of the microsatellites given the proximity of the markers to the 22q13 locus. Heterozygosity for one of these microsatellite markers then validates a true homozygous germline CYP2D6 genotype tested in the same tumor block. Patients with a homozygous CYP2D6 genotype were excluded from our CYP2D6 analysis if influence of LOH on the CYP2D6 genotype in the tumor block could not be ruled out (i.e., in case of “homozygosity” of all microsatellite markers). Further details can be found in the supplementary methods section.
Statistical analysis
For comparison of proportions and means, χ2 statistics and the Student’s t test were used. Cox regression analysis was used to assess whether DFS differed with respect to age at diagnosis, surgical procedure, tumor size, grade, nodal status, adjuvant chemotherapy or radiotherapy, and the CYP2D6 genotypes or phenotypes. In an additional exploratory analysis, 24 genetic variants of other metabolic enzymes and the estrogen receptor were associated with DFS. Statistical analyses were performed using SPSS 20.0 (SPSS Inc., Chicago, IL). Covariates were included in the multivariable model if they were of clinical significance (tumor size, nodal status, grade, and chemotherapy) or had a univariable P-value < 0.1. Genetic variants were initially tested in a general model (2 degrees of freedom). If this test resulted in a P-value < 0.1, the genetic variant was fitted and the most appropriate model (gene–dose, dominant or recessive) was selected. The distributions of DFS were estimated overall using the Kaplan–Meier method. A log-rank test was used to assess the association between the genetic variant and the outcome of interest. All results from the multivariable Cox regression analysis with a P-value < 0.05 were considered significant. No correction for multiple testing for the 24 genetic variants other than CYP2D6 was applied, since this was an exploratory analysis.
Results
Tumor blocks were collected from 746 patients enrolled in the TEAM trial and randomized to tamoxifen followed by exemestane from 59 of the 69 Dutch hospitals. Fifteen patients were ineligible because of distant metastasis at diagnosis (n = 1), an ER/PgR negative primary tumor (n = 4), a history of previous breast cancer (n = 8) and because the patient never started tamoxifen therapy (n = 2). The primary analysis therefore was performed on the 731 eligible patients. Twenty-nine genetic variants were successfully genotyped using Taqman assays except for CYP2D6*3 which was genotyped with pyrosequencing (Table 1) [28]. Genotype frequencies of 10 selected genetic variants showed deviation from Hardy–Weinberg equilibrium, but were still considered appropriate to analyze, because they did not differ from the frequencies previously reported in literature or on the NCBI website (Table 1). In Table 2, the 731 eligible patients are described. These patients were similar to the whole group of Dutch patients randomized to the sequential arm of tamoxifen followed by exemestane (n = 1,379) with regard to age, surgery, tumor size, grade, nodal status, adjuvant chemotherapy, and radiotherapy (P > 0.05, data not shown). The total number of DFS-t events in the 731 patients was 60 with a median follow-up time of 2.5 years until tamoxifen discontinuation. 25.3 and 59.1 % of patients received adjuvant chemotherapy and radiotherapy, respectively (Table 2). We translated the data from the 5 CYP2D6 alleles (*3, *4, *6, *14, *41) and concomitant CYP2D6 inhibitor use to a predicted poor, intermediate, or extensive metabolizer phenotype. Of note, ultrarapid metabolizers could not be defined since it was not possible to detect gene duplication on this source of DNA. Analysis of the CYP2D6 alleles and the three microsatellite markers D22S276, D22S2284, and D22S423 flanking the CYP2D6 gene demonstrated heterozygosity for at least one of the CYP2D6 alleles or microsatellite markers in 97.7 % of patients with a specified CYP2D6 phenotype. The 14 patients (2.3 %) with a homozygous CYP2D6 genotype in which no heterozygosity could be demonstrated for the microsatellite markers (because of homozygosity or test failure) were excluded from the analysis. The separate CYP2D6 alleles (most commonly *4 and *41) and the CYP2D6 phenotypes (poor vs extensive metabolizers: unadjusted hazard ratio 1.33, 95 % CI 0.52–3.43; P = 0.55) were not associated with DFS-t (Fig. 2a–c and Table 3). Including the 14 patients in the analysis did not alter these results (Fig. 3 supplementary files).


Kaplan Meier probabilities for disease-free survival during tamoxifen use (DFS-t) of: a CYP2D6*4 genotypes (patients excluded if influence of LOH on assigned genotype cannot be ruled out); b CYP2D6*41 genotypes (patients excluded if influence of LOH on assigned genotype cannot be ruled out); c predicted CYP2D6 phenotypes based on detection of *3, *4, *6, *14, *41 alleles and concomitant CYP2D6 inhibitor use (patients excluded if influence of LOH on assigned genotype cannot be ruled out); d ESR1 PvuII genotypes; e CYP2C19*2 genotypes; f CYP2C19*2 genotypes grouped according to a dominant model; g UGT2B15*2 genotypes; h UGT2B15*2 genotypes according to a recessive model. LOH loss of heterozygosity, DF disease free, Vt variant type allele, Wt wild type allele, PM poor metabolizer, IM intermediate metabolizer, EM extensive metabolizer
In the exploratory multivariable analysis UGT2B15*2 (Vt/Vt + Wt/Vt vs. Wt/Wt: HR 0.47, 95 % CI 0.25–0.89; P = 0.019) and ESR1-PvuII (gene–dose effect Wt/Wt > Wt/Vt > Vt/Vt: HR 1.63, 95 % CI 1.04–2.54; P = 0.033) seemed associated with DFS (Table 3). In the sensitivity analysis using the complete disease-free survival (DFS-c) as was used in the core TEAM trial analysis, CYP2D6 genotypes and phenotypes were also not associated (data not shown). In the exploratory analyses, only CYP2C19*2 (Vt/Vt vs. Wt/Vt + Wt/Wt: HR 2.39, 95 % CI 1.03–5.54; P = 0.043) was associated with DFS-c in multivariable analysis. In contrast, the more frequent CYP2C19*17 ultrarapid metabolizer allele was neither associated with DFS-t nor DFS-c.
Discussion
No association between the CYP2D6 genotypes or phenotypes and tamoxifen efficacy was found in our study. Our findings are in line with the data of two studies recently published in the Journal of the National Cancer Institute in which DNA from tumor blocks was used to genotype CYP2D6 [16, 17]. These studies were criticized because LOH in tumors would have led to false interpretation of CYP2D6 genotyping results. Indeed, the study of Regan reports a higher CYP2D6*4/*4 frequency than expected in Caucasians (16). To avoid this, we performed the first CYP2D6 association study using DNA from FFPE tumor blocks in which potentially false genotypes resulting from LOH in tumor tissue were excluded. Of note, even after controlling for LOH, CYP2D6*4 and *41 still exhibited statistically significant departures from the Hardy–Weinberg Equilibrium (Table 1). The genotype frequencies in the current study however, did not significantly differ from those in a population of Dutch early breast cancer patients, enrolled in a prospective trial in which DNA derived from whole blood was used for genotyping (CYPTAM: NTR1509, unpublished data). The reason why LOH did not affect our genotyping results in the majority of patients is probably because the slices of FFPE tumor tissue from which DNA is isolated contained substantial amounts of normal tissue. Previous studies in which 100 % concordance between CYP2D6 genotype in normal and tumor tissue is demonstrated strengthen this finding [8, 29]. This however does not implicate that future genotyping studies can be validly performed using DNA from tumor blocks when the gene is known to suffer from LOH in the tumor. In tumor enriched samples LOH may still cause false interpretation of the genotyping result. Especially when genetic variants with low allele frequency are studied, a small amount of false genotyping results could notably impact the study results. Exclusion of those possibly false genotypes by using microsatellite analysis as presented here is recommended and will enable the future use of archived FFPE tumor blocks for pharmacogenetic studies, especially when genes are studied that exhibit LOH in tumor. This may be very useful because large clinical trials contain valuable clinical data but often only have FFPE tumor tissue available for genotyping [25].
Compared to other publications on heterogeneous populations the current study was performed in a trial population, with good documentation of patient data enabling a broad multivariable analysis. Concomitant CYP2D6 inhibitor use and tamoxifen adherence may interact with the CYP2D6 genotype and may be associated with clinical outcome [12, 13, 30]. Therefore, these factors may cause confounding. In this study information on concomitant medication including CYP2D6 inhibitors (e.g., paroxetine) was available, enabling a more accurate classification into CYP2D6 phenotypes. However, the low prescription frequency (1.9 %) may suggest incomplete registration [12]. To our knowledge adjustments for tamoxifen compliance have only been made in one previous report [31]. In the current TEAM trial cohort, planned and unplanned tamoxifen discontinuation dates were registered. By censoring disease-free survival at the time of tamoxifen discontinuation (DFS-t), effect modification by aromatase inhibitors is prevented and the chance of confounding by compliance is reduced. This method however limits the follow-up duration and number of events, thus decreasing the study’s statistical power. For the CYP2D6 phenotype, our study is powered (1-β = 80 %) to detect a 2.1-fold increased risk for poor and intermediate metabolizers compared to extensive metabolizers. By performing a sensitivity analysis of CYP2D6 phenotype using the complete DFS (DFS-c), not censored at the end of tamoxifen use, thus including the years on exemestane, the number of events increases from 55 to 138. In this analysis, our study is powered (1-β = 80 %) to detect a 1.6-fold increased risk for poor and intermediate metabolizers compared to extensive metabolizers. However, despite this increase in statistical power the CYP2D6 phenotype is still not associated with disease-free survival (P = 0.42). The sequential exemestane use may have decreased the detrimental effect of CYP2D6 genotype on tamoxifen efficacy. In current daily practice however, optimal endocrine therapy for postmenopausal patients includes an aromatase inhibitor which generally is given after 2.5 years of tamoxifen. Our study results therefore apply to most postmenopausal early stage breast cancer patients that are currently treated with tamoxifen.
Kiyotani suggested in a subgroup analysis that chemotherapy modifies the effect of CYP2D6 genotype on clinical outcome [32]. An association between CYP2D6 genotype and outcome was only found in patients treated with tamoxifen, but not receiving chemotherapy, in contrast to patients who also received adjuvant chemotherapy. In our study however, subgroup analyses of patients with and without chemotherapy still resulted in a null association. Another possible explanation for our findings is that the CYP2D6 genotype mostly affects the late breast cancer recurrences. As was shown by Schroth the differences in DFS between the various CYP2D6 phenotypes in patients treated with 5 years of adjuvant tamoxifen became more apparent after 5 years of follow-up [9]. Finally, the possibility of a real association between CYP2D6 genotype and tamoxifen efficacy with a smaller effect size cannot be precluded.
Although the association with CYP2D6 could not be replicated, our exploratory analyses suggest that polymorphisms in UGT2B15 and the estrogen receptor-1 impact DFS-t in breast cancer patients treated with adjuvant tamoxifen. The UGT2B15*2 and ESR1-PvuII polymorphisms are common and therefore potentially clinically relevant. The UGT2B15*2 polymorphism has been linked to decreased glucuronidation and clearance and may lead to accumulation of active metabolites and thus a better response to tamoxifen [20, 33]. In contrast, previous studies failed to show an association between UGT2B15 and clinical outcome in breast cancer patients using tamoxifen [10, 34]. In our study, a decreased DFS-t was found with increasing number of variant (C) alleles of ESR1 PvuII. The ESR1 PvuII polymorphism has been associated with different side effects of tamoxifen [35–37]. We hypothesized that polymorphisms in ESR1 may change tamoxifen efficacy by alterations in estrogen receptor binding or signaling. The ESR1 PvuII genotype has been associated with susceptibility to the effects of hormone therapy on mammographic density in postmenopausal women: increased mammographic density was observed in women on hormone replacement therapy harboring the ESR1 PvuII C/T (=Wt/Vt) and T/T genotype, but not the C/C genotype [38]. While high-mammographic density is a known risk factor for breast cancer, reduction in mammographic density is observed during tamoxifen use and therefore may be a marker for tamoxifen response in breast cancer patients [39]. Hypothetically, the ESR1 PvuII genotype may be associated with susceptibility to the effect of tamoxifen on mammographic density and breast cancer recurrence. Although this hypothesis has not been supported by published data, an association between ESR1 PvuII genotype and tamoxifen efficacy has been reported [40]. In the sensitivity analysis, UGT2B15*2 and ESR1 PvuII were not associated with the complete DFS suggesting effect modification by exemestane. Because of the exploratory nature of these analyses no adjustments for multiple testing were made, therefore these findings may also be caused by chance. In addition, LOH in tumor tissue may have influenced these results as we did not account for potential LOH in our exploratory analyses.
In conclusion, we could not detect an association between CYP2D6 genotype or phenotype and tamoxifen efficacy, which is in line with previous data from other large studies [14–17].The current study, however, is the first CYP2D6 association study in which the potential influence of LOH in tumor blocks was accounted for, justifying the use of DNA retrieved from FFPE tumor blocks. Our broader exploratory pathway analysis showed that UGT2B15*2 and ESR1-PvuII may be associated with DFS, although these findings need validation.
References
Desta Z, Ward BA, Soukhova NV, Flockhart DA (2004) Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther 310:1062–1075
Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P et al (2003) Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 95:1758–1764
Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH et al (2005) CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst 97:30–39
Lim YC, Desta Z, Flockhart DA, Skaar TC (2005) Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol 55:471–478
Lim YC, Li L, Desta Z, Zhao Q, Rae JM, Flockhart DA et al (2006) Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J Pharmacol Exp Ther 318:503–512
Wu X, Hawse JR, Subramaniam M, Goetz MP, Ingle JN, Spelsberg TC (2009) The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res 69:1722–1727
Murdter TE, Schroth W, Bacchus-Gerybadze L, Winter S, Heinkele G, Simon W et al (2011) Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin Pharmacol Ther 89:708–717. doi:10.1038/clpt.2011.27
Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW et al (2005) Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 23:9312–9318
Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S et al (2009) Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 302:1429–1436
Dezentje VO, Guchelaar HJ, Nortier JW, van de Velde CJ, Gelderblom H (2009) Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 15:15–21
Lash TL, Lien EA, Sorensen HT, Hamilton-Dutoit S (2009) Genotype-guided tamoxifen therapy: time to pause for reflection? Lancet Oncol 10:825–833
Dezentje VO, van Blijderveen NJ, Gelderblom H, Putter H, van Herk-Sukel MP, Casparie MK et al (2010) Effect of concomitant CYP2D6 inhibitor use and tamoxifen adherence on breast cancer recurrence in early-stage breast cancer. J Clin Oncol 28:2423–2429
Kelly CM, Juurlink DN, Gomes T, Duong-Hua M, Pritchard KI, Austin PC et al (2010) Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: a population based cohort study. BMJ 340:c693
Abraham JE, Maranian MJ, Driver KE, Platte R, Kalmyrzaev B, Baynes C et al (2010) CYP2D6 gene variants: association with breast cancer specific survival in a cohort of breast cancer patients from the United Kingdom treated with adjuvant tamoxifen. Breast Cancer Res 12:R64
Goetz M, Berry D, Klein T, International Tamoxifen Pharmacogenomics Consortium (2009) Adjuvant tamoxifen treatment outcome according to cytochrome P450 2D6 (CYP2D6) phenotype in early stage breast cancer: findings from the International Tamoxifen Pharmacogenomics Consortium. Cancer Res 69:33
Regan MM, Leyland-Jones B, Bouzyk M, Pagani O, Tang W, Kammler R et al (2012) CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the breast international group 1–98 trial. J Natl Cancer Inst 104:441–451. doi:10.1093/jnci/djs125
Rae JM, Drury S, Hayes DF, Stearns V, Thibert JN, Haynes BP et al (2012) CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J Natl Cancer Inst 104:452–460. doi:10.1093/jnci/djs126
Madlensky L, Natarajan L, Tchu S, Pu M, Mortimer J, Flatt SW et al (2011) Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther 89:718–725. doi:10.1038/clpt.2011.32
Gjerde J, Hauglid M, Breilid H, Lundgren S, Varhaug JE, Kisanga ER et al (2008) Effects of CYP2D6 and SULT1A1 genotypes including SULT1A1 gene copy number on tamoxifen metabolism. Ann Oncol 19:56–61. doi:10.1093/annonc/mdm434
Nishiyama T, Ogura K, Nakano H, Ohnuma T, Kaku T, Hiratsuka A et al (2002) Reverse geometrical selectivity in glucuronidation and sulfation of cis- and trans-4-hydroxytamoxifens by human liver UDP-glucuronosyltransferases and sulfotransferases. Biochem Pharmacol 63:1817–1830
Sun D, Sharma AK, Dellinger RW, Blevins-Primeau AS, Balliet R, Chen G et al (2007) Glucuronidation of active tamoxifen metabolites by the human UDP-glucuronosyltransferases (UGTs). Drug Metab Dispos 35:2006–2014
Nakamura Y, Ratain MJ, Cox NJ, McLeod HL, Kroetz DL, Flockhart DA (2012) Re: CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the Breast International Group 1–98 trial. J Natl Cancer Inst 104:1264–1268. doi:10.1093/jnci/djs304
Pharoah PD, Abraham J, Caldas C (2012) Re: CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the Breast International Group 1–98 trial and Re: CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J Natl Cancer Inst 104:1263–1264. doi:10.1093/jnci/djs312
Stanton V Jr (2012) Re: CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: the Breast International Group 1–98 trial. J Natl Cancer Inst 104:1265–1266. doi:10.1093/jnci/djs305
Wheeler HE, Maitland ML, Dolan ME, Cox NJ, Ratain MJ (2012) Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet 14:23–34. doi:10.1038/nrg3352
Castells A, Gusella JF, Ramesh V, Rustgi AK (2000) A region of deletion on chromosome 22q13 is common to human breast and colorectal cancers. Cancer Res 60:2836–2839
van de Velde CJ, Rea D, Seynaeve C, Putter H, Hasenburg A, Vannetzel JM et al (2011) Adjuvant tamoxifen and exemestane in early breast cancer (TEAM): a randomised phase 3 trial. Lancet 377:321–331
Baak-Pablo R, Dezentje V, Guchelaar HJ, van der Straaten T (2010) Genotyping of DNA samples isolated from formalin-fixed paraffin-embedded tissues using preamplification. J Mol Diagn 12:746–749
Ahern TP, Christensen M, Cronin-Fenton DP, Lunetta KL, Rosenberg CL, Sorensen HT et al (2010) Concordance of metabolic enzyme genotypes assayed from paraffin-embedded, formalin-fixed breast tumors and normal lymphatic tissue. Clin Epidemiol 2:241–246. doi:10.2147/CLEP.S13811
Rae JM, Sikora MJ, Henry NL, Li L, Kim S, Oesterreich S et al (2009) Cytochrome P450 2D6 activity predicts discontinuation of tamoxifen therapy in breast cancer patients. Pharmacogenomics J 9:258–264
Thompson AM, Johnson A, Quinlan P, Hillman G, Fontecha M, Bray SE et al (2010) Comprehensive CYP2D6 genotype and adherence affect outcome in breast cancer patients treated with tamoxifen monotherapy. Breast Cancer Res Treat 125:279–287
Kiyotani K, Mushiroda T, Hosono N, Tsunoda T, Kubo M, Aki F et al (2010) Lessons for pharmacogenomics studies: association study between CYP2D6 genotype and tamoxifen response. Pharmacogenet Genomics 20:565–568
Court MH, Hao Q, Krishnaswamy S, Bekaii-Saab T, Al-Rohaimi A, von Moltke LL et al (2004) UDP-glucuronosyltransferase (UGT) 2B15 pharmacogenetics: UGT2B15 D85Y genotype and gender are major determinants of oxazepam glucuronidation by human liver. J Pharmacol Exp Ther 310:656–665
Ahern TP, Christensen M, Cronin-Fenton DP, Lunetta KL, Soiland H, Gjerde J et al (2011) Functional polymorphisms in UDP-glucuronosyl transferases and recurrence in tamoxifen-treated breast cancer survivors. Cancer Epidemiol Biomarkers Prev 20:1937–1943
Jin Y, Hayes DF, Li L, Robarge JD, Skaar TC, Philips S et al (2008) Estrogen receptor genotypes influence hot flash prevalence and composite score before and after tamoxifen therapy. J Clin Oncol 26:5849–5854
Onitilo AA, McCarty CA, Wilke RA, Glurich I, Engel JM, Flockhart DA et al (2009) Estrogen receptor genotype is associated with risk of venous thromboembolism during tamoxifen therapy. Breast Cancer Res Treat 115:643–650
Hayes DF, Skaar TC, Rae JM, Henry NL, Nguyen AT, Stearns V et al (2010) Estrogen receptor genotypes, menopausal status, and the effects of tamoxifen on lipid levels: revised and updated results. Clin Pharmacol Ther 88:626–629. doi:10.1038/clpt.2010.143
van Duijnhoven FJ, Peeters PH, Warren RM, Bingham SA, Uitterlinden AG, van Noord PA et al (2006) Influence of estrogen receptor alpha and progesterone receptor polymorphisms on the effects of hormone therapy on mammographic density. Cancer Epidemiol Biomarkers Prev 15:462–467
Cuzick J, Warwick J, Pinney E, Warren RM, Duffy SW (2004) Tamoxifen and breast density in women at increased risk of breast cancer. J Natl Cancer Inst 96:621–628
Grabinski JL, Chisholm G, Smith LS, Drengler RL, Kalter S, Rodriguez G et al. (2008) ER alpha genotypes and breast cancer recurrence. J Clin Oncol 26(15S):501
Acknowledgments
We would like to thank the women participating in the TEAM trial and Tom van Wezel for his intellectual contribution and assistance in the microsatellite analysis. The core TEAM trial was supported by an unrestricted research grant from Pfizer Inc. The current separate pharmacogenetic study was not financially supported. Pfizer had no role in writing of this manuscript or decision to submit.
Disclosure
The authors have declared no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Dezentjé, V.O., van Schaik, R.H.N., Vletter-Bogaartz, J.M. et al. CYP2D6 genotype in relation to tamoxifen efficacy in a Dutch cohort of the tamoxifen exemestane adjuvant multinational (TEAM) trial. Breast Cancer Res Treat 140, 363–373 (2013). https://doi.org/10.1007/s10549-013-2619-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2619-6