
Abstract
To examine the role of germline genetic variations in inflammatory pathways as modifiers of time to recurrence (TTR) in patients with early stage breast cancer (BC), DNA from 997 early stage BC patients was genotyped for 53 tagging single nucleotide polymorphisms (SNPs) in 12 genes involved in inflammation. SNPs were analyzed separately for Caucasians versus African-Americans and Hispanics. Cox proportional hazards models were used to evaluate the association between SNPs in the inflammatory genes and TTR, adjusted for clinical and pathologic covariates. In univariable analyses of Caucasian women, the homozygous genotype of 12 SNPs, including 6 NFKB1 SNPs, 4 IL4 SNPs, and 2 IL13 SNPs, were significantly associated with a decrease in TTR compared with the heterozygous and/or corresponding homozygous genotype (P < 0.05). The significant NFKB1 and IL4 SNPs were in an area of high linkage disequilibrium (D′ > 0.8). After adjusting for stage, age, and treatment, carriage of the homozygous genotypes for NFKB1 rs230532 and IL13rs1800925 were independently associated with a shorter TTR (P = 0.001 and P = 0.034, respectively). In African-American and Hispanic patients, expression of NFKB1 rs3774932, TNFrs1799964, and IL4rs3024543 SNPs were associated with a shorter TTR in univariable model. Only NFKB1 rs3774932 (P = 0.02) and IL4Rrs3024543 (P = 0.03) had independent prognostic value in the multivariable model These data support the existence of host genetic susceptibility as a component in recurrence risk mediated by pro-inflammatory and immune factors, and suggest the potential for drugs which modify immune responses and inflammatory genes to improve prognosis in early stage BC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) remains the most frequent malignant neoplasm in North American women [1]. Although earlier diagnosis and new and improved treatments have changed the overall prognosis in women diagnosed with early stage BC, 30–40 % of women experience a recurrence of their cancer within 3–5 years of diagnosis.
The prognosis of early stage BC is influenced by a number of well-established factors, including tumor stage [2], axillary lymph node status [3], tumor grade [4], and pathologic markers such as estrogen receptor (ER), progesterone receptor (PR), and HER2/neu oncoprotein expression [5, 6]. Hierarchical clustering of gene expression data from tumor samples of BC patients has identified specific genes that can be used to divide patients into prognostic subgroups on the basis of expression of the above markers [7, 8]. Furthermore, specific patterns of gene expression within subgroups that include inflammatory and immune response gene signatures can further stratify patients in terms of overall and disease-free survival, irrespective of the type of therapy [9–12]. An inflammatory tumor environment can result in tumor promotion through enhanced proliferation and survival of malignant cells and the subversion of innate and adoptive immune responses (reviewed in [13]).
Several investigators have attempted to correlate germline single nucleotide polymorphisms (SNPs) of inflammatory cytokine genes with BC risk, with mixed results [14, 15]. A recent large meta-analysis showed no correlation between the expression of a large number of cytokine SNPs and the risk of developing BC [16], but only a few small studies have specifically examined whether SNPs in inflammatory genes are associated with BC prognosis (reviewed in [17]). To address this gap in knowledge, we utilized a large cohort of early stage BC patients and examined the role of 53 tagging SNPs in 12 genes involved in the inflammatory pathway in predicting time to recurrence (TTR).
Materials and methods
Study population
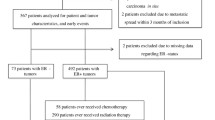
Detailed clinical information, including patient age, disease stage, nuclear grade, ER and PR status, and primary treatment including surgery, radiation, chemotherapy, and/or endocrine therapy, was retrospectively abstracted from the medical records of a cohort of 1,390 BC patients diagnosed between 1985 and 2002, who were enrolled in the Early Stage Breast Cancer Repository at the MD Anderson Cancer Center. [18]. Of these 1,390 patients, 1,089 had blood or normal lymph node samples available for genotyping. Fifty-nine patients were excluded from study analysis because of assay failure (n = 54), insufficient clinical information (n = 5), or lack of information on race (n = 33). The study analysis included 997 patients (739 Caucasians, 141 African-Americans, and 117 Hispanics). The study was approved by the Institutional Review Board of MD Anderson Cancer Center.
SNP selection and genotyping
We reviewed the existing literature and selected 53 SNPs from 12 genes associated with inflammation in BC (IL6, IL8, TNF, IL4, IL4, IL4R, IL13, PTGS2 [COX2], TGFB1, IL10, IL1B, and IL1RN). All selected SNPs were within genes or linkage disequilibrium (LD) blocks containing these genes and were identified using data from the HapMap Project (www.hapmap.org, version 23). All SNPs met the following criteria: minor allele frequency of ≥0.05, Illumina design score >0.4, and r 2 ≥ 0.8 for binning. We genotyped samples using Illumina Golden Gate technology (San Diego, CA, USA) as part of a larger array of 1,514 SNPs in total. Genomic DNA was extracted from blood or buccal samples using the QIAamp DNA Blood Maxi kit (QIAGEN) and from paraffin-embedded (FFPE) nodal tissue using the PicoPure DNA extraction method (Applied Biosystems, Foster City, CA, USA). The call rates from the FFPE samples and blood or buccal samples were 92 and 99 %, respectively. Blinded duplicate samples were included in the array platform and the duplication concordance was 100 %. All genotyping clustering was performed using GenomeStudio data analysis software (Illumina).
Statistical analyses
We analyzed SNPs associated with TTR in Caucasian separately from African-American and Hispanic patients because of racial differences in the frequency of SNPs involving multiple genes. TTR was defined as the time between the date of first treatment and date of BC loco-regional or distant metastatic recurrence. The relationship between each SNP and clinical variable and TTR was evaluated separately using a univariate Cox proportional hazards regression model. [19]. To build a Cox proportional hazards model with high-dimensional covariates, we applied the CoxBoost algorithm to the SNPs that had prognostic potential suggested by the univariable analyses (P ≤ 0.05) and then used a backwards elimination procedure (α = 0.05) [20]. Interaction terms between significant SNPs from univariable analyses and hormone receptor status (ER+ or PR+ vs. ER− and PR−) were constructed to test for interaction among Caucasian patients. Interactions between SNPs and hormone receptor status were assessed using the likelihood-ratio test (P < 0.05) comparing the model including an interaction term with the reduced model without the interaction term adjusted for age, stage, and treatment. Stratified multivariable analysis was performed where effect modification was observed. Linkage mapping was performed using Haploview version 4.2 [21] for the LD blocks, assuming 1 Mb = 1 cM, using the SNPs which had prognostic significance in the univariable model. All other analyses were conducted using SAS version 9.1 (SAS Institute, Cary, NC).
Results
Univariable analysis of clinical and pathologic variables and breast cancer recurrence
There were 182 recurrences with a median time to follow-up of 18 years. Table 1 lists the clinical and pathologic covariates associated with TTR in univariable analysis. Variables significantly associated with a prolonged TTR included clinical stage I disease, ER or PR positivity, patient age ≥50 years, and use of hormonal therapy alone.
Univariable analyses of SNPs associated with TTR in Caucasian women
In the univariable analysis of the 53 SNPs in 739 Caucasian patients, 6 SNPs of NFKB1, 4 SNPs of IL4, and 2 SNPs of IL13 were associated with significant differences in TTR (P < 0.05). After multiple testing adjustments those 12 SNPs were significant at a false discovery rate (FDR) of 0.12 (Table 2). As shown in Fig. 1, there was significant LD between the NFKB1 SNPs (SNPs 1–6, Fig. 1). Similarly, the four IL4 SNPs (9–12, Fig. 1) were in a block of high LD, but LD was not demonstrated for IL13 SNPs rs1800925 and rs1295686.

Linkage analysis of NFKB1 and IL4 SNPs found to be prognostically significant in univariable analysis. All of the NFKB1 SNPs as well as the four IL4 SNPs were in a single block of LD (D′ > 0.9)
Multivariable analyses of SNPs associated with TTR in Caucasian women
In multivariable analysis, SNPs NFKB1 rs230532 (IVS2-910G>A) and IL13 rs1800925 (−1069C>T), were independent predictors of a shorter TTR adjusted for age, stage, and treatment (Table 3). There was evidence of a significant interaction between SNPs NFKB1 rs3774932 (P interaction = 0.02) and rs230532 (P interaction = 0.01) and hormone receptor status. In a stratified multivariable model adjusted for age, stage, and treatment, carriers of the SNPs NFKB1 rs230532 (TT vs. AA+TT) and NFKB1 rs3774932 (AA vs. GG) genotypes had a shorter TTR only in the ER+ or PR+ subgroup (Table 4).
Prognostic impact of SNPs in non-Caucasian women
To determine if the frequency and prognostic impact of inflammatory SNPs found in Caucasians were the same or different as those in non-Caucasian patients, we performed similar analyses using 53 SNPs in the 141 African-American and 117 Hispanic patients combined (Tables 5, 6). Univariable analysis revealed that carriage of the less frequent homozygous genotype of NFKB1 rs3774932, was associated with a significantly shorter TTR (P = 0.03) in these ethnic groups (Table 5). A significant trend for shorter TTR was observed for two additional polymorphisms not found in Caucasians, TNF rs1799964 (P = 0.07) and IL4R rs3024543. (P = 0.09). Multivariable analysis including the three SNPs that were associated with TTR in the univariable model and adjusted for stage and hormonal therapy revealed that only SNPs NFKB1 rs3774932 (P = 0.02) and IL4R rs3024543 (P = 0.03) were independent predictors for shorter TTR (Table 6).
Discussion
To our knowledge, this retrospective study is one of the first to examine the prognostic impact of 53 germline polymorphisms from 12 inflammatory genes in a large well-characterized homogeneous cohort of patients with early stage BC. After adjusting for prognostic clinical parameters, NFKB1 rs230532 and IL13 rs1800925 SNPs were independent predictors of a shorter TTR in Caucasians, while NFKB1 rs3774932 and IL4R rs3024543 SNPs were predictive of a shorter TTR in African-Americans and Hispanics.
NFKB encodes nuclear factor kappa B (NFkB) a family of proteins consisting of five hetero-dimeric transcription factors [22, 23] which serve as a master regulators for a plethora of genes involved in inflammation, cell proliferation, apoptosis inhibition, bone remodeling angiogenesis, chemokine production, and metalloproteinase production [24, 25]. Despite the abundance of information in regards to the function of NFkB in tumors, there are no published studies assessing the putative role of NFKB germline polymorphisms and prognosis in BC. A study by Kurt et al. [26] demonstrated that peripheral T lymphocytes from women with BC demonstrated an impaired ability to translocate NFkB p65 (Rel-A) following activation by anti-CD3 and Interleukin-2 (IL2).
We were unable to evaluate the association between SNP expression and TTR by molecular subtypes (8) because patients in our BC cohort were treated between 1985 and 2000, prior to the routine assessment of HER2/neu status. However, when stratified by ER and PR receptor status, Caucasian patients with ER+ or PR+ disease had a shorter TTR if they carried NFKB1 genotypes rs230532 and rs3774932. This is an interesting finding in view of the fact that inflammation in general has been shown to be associated with more aggressive ER+BC [27]. Moreover, it has recently been demonstrated that NFkB activation can maintain immunosuppressive function of type 2 tumor-associated macrophages (TAM) [28] which have been found to comprise up to 50 % of breast tumor mass [29]. Although NFkB activation has been demonstrated more frequently in ER negative tumors [30], recent evidence has demonstrated antagonistic cross-talk between the NFkB and ER pathways [31]. Additional evidence suggests that NFkB activation occurs in a subset of ER+ tumors and cell lines with poor response to endocrine therapy and that incubation of an NFkB inhibitor with resistant lines could restore endocrine sensitivity [32, 33] Hence, our findings may have relevance with respect to predicting those ER+ patients who are resistant to endocrine therapy.
As confirmed by this study, variations in allele frequencies and specific SNPs associated with inflammation have been observed for different races [34]. Erdei et al. [35] measured polymorphisms in cytokine genes along with secreted cytokines in New Mexican Hispanic women with BC compared to age-, gender-, and smoking-matched women without incident BC (controls) and found a higher frequency of SNPs rs2069705, rs2243248, and rs1800925 located in the promoter regions of the interferon gamma (IFNγ), IL4 , and IL13 genes, respectively. The authors hypothesized that immune dysregulation might account for more aggressive tumors found in Hispanics compared to Whites.
IL13 and IL4 are members of the TH2 cytokine family along with IL10, IL6, and TGFβ, and are involved in immune suppression, antibody production, and activation of other pro-inflammatory molecules [36]. IL4 and IL13 are crucial immune factors which signal through the IL4 receptor (IL4R). A recent large prospective study in patients with diffuse large B cell lymphoma demonstrated a favorable impact of the interleukin-4 receptor allelic variant I75 (rs1805010) on overall survival [37]. Preclinical studies in mice have shown that CD4-positive T cells infiltrating breast tumors secrete high levels of IL13, which promotes tumor growth through immune suppression by dendritic cells, an effect that could be prevented by administration of IL13 antagonists [38].
There was close linkage between all of the significant NFKB1 and IL4 SNPs in Caucasians. Most of these SNPs were intronic, except for IL4 SNP rs2070874 which was located near the 5′ UTR. Hence the latter SNP is more likely to influence transcription, whereas the mechanisms of the intronic SNP are less clear. It is more likely that either SNP could be in LD with another functional SNP in the NFKB1or IL4 gene region or that one or more of the SNPs could be in some other regulatory region of the gene. Preclinical studies suggest that the presence of SNPs in the regulatory or coding regions of cytokine genes can result in functional alterations in the transcriptional regulation of these genes or the proteins they encode [39]. Dominant homozygous polymorphisms within the promoter regions of their respective genes induce higher circulating levels of cytokines and other growth factors, resulting in increased oncogenesis and inflammation resulting in immune suppression [40].
In univariable analysis, there was a significant trend (P = 0.07) for carriage of TNF SNP rs1799964 and a shorter TTR in African-American and Hispanic patients. This SNP was not an independent predictor of prognosis when stratified for other clinical covariates in the multivariable model. The product of the TNF gene, TNFα, is a pro-inflammatory cytokine induced under hypoxic conditions [41]. Chronic production of TNFα in benign and malignant BC tissues is associated with a poor outcome [42]. Previous investigators have demonstrated that carriage of several TNF SNPs significantly correlated with shorter disease-free survival and overall survival [43, 44]. In one study, The TNF type II homozygous genotype correlated with higher levels of TNFα in serum and a worse prognosis than the TNF type I genotype [44]. Whether the SNP rs1799964 identified in our study is associated with increased serum levels of TNFα is currently unknown and needs further study.
A significant weakness of our study was that serum cytokine levels were not measured, making it difficult to associate specific cytokine SNPs with immune function. In a case control study, 13 cytokines were measured in 40 New Mexican Hispanic breast cancer patients compared to 40 controls (34). Of the 13 cytokines observed, only 5: IL1β, IL-5, TNFα, IL6, and IL2 showed elevated levels in BC patients compared to controls. A significant problem with cytokine measurements in association with disease states is that levels can vary significantly based on the response to chemotherapy [45], and other factors associated with inflammation. Additional large studies which assess SNP expression with cytokine function in different ethnic groups with early stage BC are needed.
Another possible weakness is that the low recurrence rate along with stratification by race and other variables may have resulted in our study being underpowered. Larger cohort studies that allow for the evaluation of breast cancer molecular subtypes and interactions between polymorphisms and other clinical and environmental factors are needed to validate and extend our findings. Despite these limitations, our data, as well as data from several smaller studies has demonstrated that the presence of specific polymorphisms of genes for molecules and cytokines involved in inflammation and tumor immunity may have prognostic value in BC patients. These findings could have important implications for the use of blocking antibodies and other molecules that target the protein products generated through over expression of the significant gene polymorphisms involved [46] in an attempt to improve the disease-free survival of patients with early stage BC.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ (2009) Cancer statistics, 2009. CA Cancer J Clin 59(4):225–249
Brewster AM, Hortobagyi GN, Broglio KR, Kau SW, Santa-Maria CA, Arun B et al (2008) Residual risk of breast cancer recurrence 5 years after adjuvant therapy. J Natl Cancer Inst 100(16):1179–1183
Cianfrocca M, Goldstein LJ (2004) Prognostic and predictive factors in early-stage breast cancer. Oncologist 9(6):606–616
Le Doussal V, Tubiana-Hulin M, Friedman S, Hacene K, Spyratos F, Brunet M (1989) Prognostic value of histologic grade nuclear components of Scarff–Bloom–Richardson (SBR). An improved score modification based on a multivariate analysis of 1,262 invasive ductal breast carcinomas. Cancer 64(9):1914–1921
Hess KR, Pusztai L, Buzdar AU, Hortobagyi GN (2003) Estrogen receptors and distinct patterns of breast cancer relapse. Breast Cancer Res Treat 78(1):105–118
Menard S, Fortis S, Castiglioni F, Agresti R, Balsari A (2001) HER2 as a prognostic factor in breast cancer. Oncology 61(Suppl 2):67–72
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K et al (2004) Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2(2):E7
Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C (2007) An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol 8(8):R157
Yau C, Esserman L, Moore DH, Waldman F, Sninsky J, Benz CC (2010) A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res 12(5):R85
Bianchini G, Qi Y, Alvarez RH, Iwamoto T, Coutant C, Ibrahim NK et al (2010) Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol 28(28):4316–4323
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454(7203):436–444
Le Marchand L, Haiman CA, van den Berg D, Wilkens LR, Kolonel LN, Henderson BE (2004) T29C polymorphism in the transforming growth factor beta1 gene and postmenopausal breast cancer risk: the Multiethnic Cohort Study. Cancer Epidemiol Biomarkers Prev 13(3):412–415
Smith KC, Bateman AC, Fussell HM, Howell WM (2004) Cytokine gene polymorphisms and breast cancer susceptibility and prognosis. Eur J Immunogenet 31(4):167–173
Balasubramanian SP, Azmy IA, Higham SE, Wilson AG, Cross SS, Cox A et al (2006) Interleukin gene polymorphisms and breast cancer: a case control study and systematic literature review. BMC Cancer 14(6):188
Kim S, Hagemann A, DeMichele A (2009) Immuno-modulatory gene polymorphisms and outcome in breast and ovarian cancer. Immunol Invest 38(3–4):324–340
Brewster AM, Do K-A, Thompson PA, Hahn KM, Sahin AA, Cao Y, Stewart MM, Murray JL, Hortobagyi GN, Bondy ML (2008) The relationship between epidemiological risk factors and breast cancer recurrence. J Clin Oncol 25(28):4438–4444
Pounds S, Morris SW (2003) Estimating the occurrence of false positive and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics 19:1236–1242
Binder H, Schumacher M (2008) Allowing for mandatory covariates in boosting estimation of sparse high-dimensional survival models. BMC Bioinformatics 9:1–10
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265
Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109(Suppl):S81–S96
Nakshatri H, Goulet RJ Jr (2002) NF-kappaB and breast cancer. Curr Probl Cancer 26(5):282–309
Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE et al (2004) Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306(5696):704–708
Barkett M, Gilmore TD (1999) Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18(49):6910–6924
Kurt RA, Urba WJ, Smith JW, Schoof DD (1998) Peripheral T lymphocytes from women with breast cancer exhibit abnormal protein expression of several signaling molecules. Int J Cancer 78:16–20
Baumgarten SC, Frasor J (2012) Minireview: inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol 26(3):360–371
Hageman T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR (2008) “Re-educating” tumor-associated macrophages by targeting NF-kB. J Exp Med 206:1261–1268
Kelly PM, Davison RS, Bliss E (1988) McGee J0. Macrophages in human breast disease: a quantitative immunohistochemical study. Br J Cancer 57:174–177
Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB et al (2004) NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci USA 101(27):10137–10142
Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Sledge GW (1997) Constitutive activation of NF-kB during progression of breast cancer to hormone-independent growth. Mol Cell Biol 17:3629–3639
de Graffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, Freeman JW, Weiss GR (2004) NF-kB inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to Tamoxifen. Ann Oncol 15:885–890
Riggins RB, Zwart A, Nehra R, Clarke R (2005) The nuclear factor kB inhibitor parthenolide restores ICI 182,780 (Fasolodex, fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther 4:33–41
Van Dyke AL, Cote ML, Wenzlaff AS, Land S, Schwartz AG (2009) Cytokine SNPs: comparison of allele frequencies by race and implications for future studies. Cytokine 46(2):236–244
Erdei E, Kang H, Meisner A, White K, Pickett G, Baca C, Royce M, Berwick M (2010) Polymorphisms in cytokine genes and serum cytokine levels among New Mexican Women with and without breast cancer. Cytokine 51:18–24
De Nardo DG, Coussens LM (2007) Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res 2(4):212
Schoof N, von Bonin F, Zeynalova S, Ziepert M, Jung W, Loeffler M, Pfreundschuh M, Trumper L, Kube D (2009) Favorable impact of the interleukin-4 receptor allelic variant I75 on the survival of diffuse large B-cell lymphoma patients demonstrated in a large prospective clinical trial. Ann Oncol 20(9):1548–1554
Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D et al (2007) Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med 204(5):1037–1047
Martin AM, Athanasiadis G, Greshock JD, Fisher J, Lux MP, Calzone K et al (2003) Population frequencies of single nucleotide polymorphisms (SNPs) in immuno-modulatory genes. Hum Hered 55(4):171–178
Haukim N, Bidwell JL, Smith AJ, Keen LJ, Gallagher G, Kimberly R et al (2002) Cytokine gene polymorphism in human disease: on-line databases, supplement 2. Genes Immun 3(6):313–330
Guida E, Stewart A (1998) Influence of hypoxia and glucose deprivation on tumour necrosis factor-alpha and granulocyte-macrophage colony-stimulating factor expression in human cultured monocytes. Cell Physiol Biochem 8(1–2):75–88
Miles DW, Happerfield LC, Naylor MS, Bobrow LG, Rubens RD, Balkwill FR (1994) Expression of tumour necrosis factor (TNF alpha) and its receptors in benign and malignant breast tissue. Int J Cancer 56(6):777–782
Jung JH, Chae YS, Moon JH, Kang BW, Kim JG, Sohn SK, Park JY, Lee MH, Park HY (2010) TNF superfamily gene polymorphism as prognostic factor in early breast cancer. J Cancer Res Clin Oncol 136(5):685–694
Mestiri S, Bouaouina N, Ahmed SB, Khedhaier A, Jrad BB, Remadi S et al (2001) Genetic variation in the tumor necrosis factor-alpha promoter region and in the stress protein hsp70-2: susceptibility and prognostic implications in breast carcinoma. Cancer 91(4):672–678
Pusztai L, Mendoza TR, Reuben JM, Martinez MM, Willey JS, Lara J, Syed A, Fritsche HA, Bruera E, Booser D, Valero V, Arun B, Ibrahim N, Rivera E, Royce M, Cleeland CS, Hortobagyi GN (2004) Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine 25(3):94–102
Demaria S, Pikarsky E, Karin M, Coussens LM, Chen YC, El-Omar EM et al (2010) Cancer and inflammation: promise for biologic therapy. J Immunother 33(4):335–351
Acknowledgments
The authors wish to acknowledge Ricardo Mottu and Katharina Alvarez for manuscript preparation. Supported by Grants R03 CA123553 (A.M.B.), Susan Komen Disparities Career Award (A.M.B), R01 CA089608 (M.L.B., P.T.), K07 CA160753 (M.P.), and CA16672 (MD Anderson Cancer Center) from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services.
Conflict of interest
The authors indicated no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Murray, J.L., Thompson, P., Yoo, S.Y. et al. Prognostic value of single nucleotide polymorphisms of candidate genes associated with inflammation in early stage breast cancer. Breast Cancer Res Treat 138, 917–924 (2013). https://doi.org/10.1007/s10549-013-2445-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2445-x