
Abstract
We recently reported that a ratio of high B cell and low IL-8 metagene expression identified 32 % of triple negative breast cancers (TNBC) with good prognosis and was the only significant predictor in multivariate analysis including routine clinicopathological variables. However, the clinical relevance of this signature in other breast cancer subtypes remains unclear. We compiled Affymetrix gene expression datasets from 4,467 primary breast cancer samples and excluded 329 triple negative samples which were used as discovery cohort in our previous study. Molecular classification of the remaining 4,138 samples was performed by two methods, including single genes (ER, PgR, HER2, and Ki67) and a centroid-based method using the intrinsic gene list. The prognostic value within the respective subtypes was assessed by analyzing the event-free survival of patients as a function of the B cell/IL-8 metagene ratio using previously published cutoff. ER-negative subtypes had the highest expression of the B cell and the IL-8 metagenes. The IL-8/B cell signature assigned a considerable fraction of samples (range 20.7–42.0 %) into the “good prognosis” group. However, a significant prognostic value was only observed in the subgroup of triple negative breast cancer (P = 0.035). The prognostic value of the B cell/IL-8 ratio is mainly confined to the basal-like and TNBC subtypes of breast cancer. This result underlines the importance of subtype-specific analyses and suggests a sequential multistep approach to developing and applying outcome predictors in the clinic.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is a heterogeneous disease of different molecular subtypes. Currently, the most simple and applicable stratification of breast cancer is based on expression of the hormone receptors for both estrogen (ER) and progesterone (PgR) as well as the human epidermal growth factor receptor 2 (HER2) [36]. Based on these three receptors, tumors are characterized as hormone receptor positive, HER2 positive (i.e., amplification or overexpression of HER2), or triple negative breast cancer (TNBC) lacking the expression of all three receptors. In addition, several refined stratifications applying genomic methods or the inclusion of additional immunohistochemical markers (e.g., Ki67) allow the distinction of “Basal-like” breast cancers as well as “Luminal A” and “Luminal B” subgroups each with different prognosis and clinical behavior [27–29]. In recent years, it became increasingly clear that the subtype composition of a dataset can strongly influence the prognostic and predictive gene signatures derived from it [36, 39]. Often these “first generation” signatures represent a surrogate marker for the subtype distinction itself [29]. As a consequence, several recent guidelines have suggested to analyze subtypes of breast cancers separately and to derive subtype-specific genomic tests [12, 19].
In previous work, we had assembled a large dataset of TNBC and used an unsupervised method to derive signatures which are capable of delivering highly significant prognostic information within the TNBC subgroup [31]. In the present study, we analyzed whether this signature has also prognostic value within other subtypes of breast cancer. Interestingly, we could demonstrate that despite the signature identified similar fractions of samples among the different subtypes its prognostic significance was restricted to triple negative and basal-like breast cancer. Our results underline the importance of subtype-specific analyses and suggest a sequential multistep approach for application of future genomic tests in the clinic.
Methods
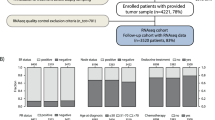
All analyses were performed according to the “REporting recommendations for tumour MARKer prognostic studies” (REMARK) [24, 35] and the respective guidelines to microarray-based studies for clinical outcomes [7]. A diagram of the complete analytical strategy and the flow of patients through the study, including the number of patients included in each stage of the analysis, is given in Fig. 1. We compiled Affymetrix gene expression data (U133A or U133Plus2.0 arrays) of 4,467 breast cancer patients from 40 publicly available datasets (Supplementary Table S1). However, 329 triple negative cases of these 4,467 samples have been used before as a finding cohort in our previous studies [17, 31]. Thus, these samples had to be excluded from all subsequent analyses resulting in 4,138 applicable samples. Affymetrix CEL files were processed with the MAS5.0 algorithm of the affy package [9] of the Bioconductor software project [10]. Data from each array were log2-transformed, median-centered, and the expression values of all the probesets from the U133A array were multiplied by a scale factor S so that the magnitude (sum of the squares of the values) equals one.

Diagram of the analytical strategy according to REMARK criteria. The analytical strategy and the flow of patients through the study is presented as recommended by the REMARK criteria [14]
To identify the intrinsic subtypes of breast cancer, we used two alternative previously described approaches: First, we used the simple method according to Hugh et al. [14] which is based on the expression of single marker genes (ESR1, PgR, HER2, and Ki67) to define TNBC-, HER2-, Luminal A-, and Luminal B-subtypes. The cutoff values for ESR1, PgR, and HER2 have been described previously [16]. For a distinction of Luminal A and Luminal B subgroups, all 2,884 ERpositive/HER2negative samples were selected and a median split according to Ki67 expression was performed. In addition, 106 ERpositive/HER2positive cases were assigned to the Luminal B subtype according to Hugh et al. [14]. As a second, alternative method for subtype determination, we applied a single sample predictor (SSP) [38] according to the centroid method using the gene set from Hu et al. [13]. The centroid analyses were performed separately in six larger datasets encompassing a total of 1,142 samples. However, 195 of those samples were used as finding cohort of triple negative cases in our previous studies [17, 31] and thus had to be excluded from further analyses presented here. This exclusion results in a reduced proportion of basal-like tumors among the remaining cohort of 947 samples (see Table 1). The individual assignments according to the two different methods of molecular subtype definition are given for each sample in Supplementary Table S2.
We have previously reported an unsupervised analysis identifying metagenes that distinguish molecular subsets within TNBC [31]. In the present study, we calculated expression values for two of these metagenes, namely the B cell and IL8 metagenes, for all 4,467 samples as the mean expression of four (IL8 metagene) and 48 (B cell metagene) Affymetrix probesets, respectively. The probesets used are listed in Supplementary Table S3. We had also reported in our previous study that high expression of the B cell metagene was associated with good prognosis and high expression of the IL-8-related metagene with poor prognosis in TNBC. We had then combined the two metagenes in the previous study into a signature: Samples with both high B cell metagene and low IL-8 metagene expression were characterized as “Good Prognosis”; all the other samples were characterized as “Poor Prognosis.” The respective cutoff values (B cell metagene >0.005 and IL-8 metagene <−0.001) were defined in a discovery cohort of 394 TNBC [31]. In the present study, we now used these pre-defined cutoff values to assess the prognostic value of the B Cell/IL-8 signature within different molecular subtypes (see Supplementary Table S2 for the assignment of each individual sample). We then analyzed the event-free survival of patients according to this signature separately in the different molecular subtypes.
In the conduct of the presented analysis, event-free survival (EFS) was calculated as preferentially corresponding to the RFS endpoint, but measured with respect to the DMFS endpoint if RFS was not available. All results from survival analyses were verified by examining the effect of the different endpoints in stratified analyses. Followup data for those women in whom the envisaged end point was not reached were censored as of the last followup date or at 120 months. Subjects with missing values were excluded from the analyses. We constructed Kaplan–Meier curves and used the log-rank test to determine the univariate significance of the variables. A Cox proportional-hazards model was used to simultaneously examine the effects of multiple covariates on survival. The effect of each individual variable was assessed with the use of the Wald test and described by the hazard ratio with a 95 percent confidence interval (95 % CI).
Results
Compilation of Affymetrix microarray datasets
Figure 1 presents the analytical strategy and the flow of patients through the study. We compiled Affymetrix gene expression data of 4,467 breast cancer patients from 40 publicly available datsets (see Methods section and Supplementary Table S1). 329 triple negative cases of these samples had to be excluded since they were used in our previous studies as a finding cohort [17, 31] leaving 4,138 applicable samples for the analyses. Table 1 gives the clinical characteristics of all 4,467 samples and of the cohort of 4,138 samples used in the subsequent analyses.
Both B cell and IL-8 metagenes display highest expression in ER negative breast cancers
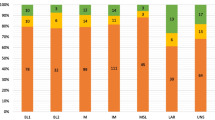
We first compared the expression of B cell and IL-8 metagenes among the different molecular subtypes of breast cancer. Molecular subtyping was performed by two alternative strategies as described in the Methods section: Either a single marker method according to Hugh et al. [14] or the centroid method applying a single sample predictor (SSP) [13, 38]. Results of the single marker method were available for all 4,138 samples: data from the centroid method for 947 cases. When the single marker method was used for stratification, we detected the lowest expression of both metagenes among the ER-positive Luminal subtypes, whereas high expression was observed in the ER negative subtypes, i.e., TNBC and HER2 (Fig. 2a, c). Similarly, when applying the centroid method for stratification, we detected the highest levels of both the IL-8 metagene and the B cell metagene in the basal-like subgroup (Fig. 2b, d). Again, the lowest expression of both metagenes was seen in the Luminal A and Luminal B subgroups (Fig. 2b, d).

Box plots of IL-8 and B cell metagene expression among different molecular subtypes of breast cancer. Expression of the IL-8 metagene is shown in panels (a) and (b), while expression of the B cell metagene is shown in panels (c) and (d). In a and c 4,138 breast cancer samples were stratified into molecular subtypes using single marker genes according to the method of Hugh et al. [14]. In panels b and d, a single subtype predictor (SSP) applying the centroid method was used to classify 947 samples from six larger dataset
Prognostic value of the IL-8/B cell signature depends on the ER status of the tumor
Since both B cell and IL-8 metagenes displayed higher expression in ER-negative disease, we next analyzed the prognostic value of the combined IL-8/B cell signature relation to the ER status of the tumor. The previously defined cutoff values were used to stratify patients in a “good prognosis group” characterized by both high B cell and low IL-8 expression and a “poor prognosis group” encompassing all the remaining samples. Followup information was available for 2,353 samples. 28.9 and 33.5 % of the samples were assigned a “good prognosis” in the ER-positive and ER-negative subgroup, respectively. This roughly similar relative proportion (about one third) of good prognosis samples in both ER-positive and ER-negative disease results from the reduced expression of both of the metagenes among ER-positive tumors (see Fig. 2). However, as shown in Fig. 3, the prognostic value of the signature is clearly dependent on ER status. We observed no prognostic value among ER-positive tumors (P = 0.36), while it was highly significant for ER negative disease (P = 0.039).

Prognostic value of the IL-8/B cell signature in ER-positive and ER-negative breast cancer. For 2,353 of the 4,138 samples, followup information was available (Table 1). The IL-8/B cell signatures were used to classify those samples into “good prognosis” or “poor prognosis.” Kaplan–Meier analyses of the samples are shown separetely for 1,941 ER-positive samples in (a) and 412 ER-negative samples in (b). A significant difference in survival was only observed in the ER negative subgroup (P = 0.039)
Prognostic value of the IL-8/B cell signature in the different molecular subtypes of breast cancer
For a more detailed analysis, we further stratified the samples according to the molecular subtypes of breast cancer. Again, we alternatively applied the two different methodologies for subtyping as given above. As shown in Table 2, in all subgroups, a considerable fraction of the samples (range 20.7–42.0 %) was assigned to “good prognosis” by the IL-8/B cell signature. We then analyzed the followup of the patients separately for the different subtypes. As demonstrated in Figs. 4 and 5, a significant prognostic value of the IL8/B cell signature was only observed in the TNBC subgroup (P = 0.035, Fig. 4a) and a strong trend in the Basal-like subtype (P = 0.061, Fig. 5a), respectively. Among all other subtypes, the IL-8/B cell signature displayed no significant prognostic value (Table 3).

Prognostic value of the IL-8/B cell signature in different molecular subtypes of breast cancer according to the single marker method. The IL-8/B cell signature was used to classify 2,353 samples with followup information as “good prognosis” or “poor prognosis.” Separate Kaplan–Meier analyses are given in panels a–d for the four different subtypes of breast cancer stratified applying the single marker method. A significant prognostic value of the IL-8/B cell signature was only observed among triple negative breast cancers (panel a, P = 0.035)

Prognostic value of the IL-8/B cell signature in different molecular subtypes of breast cancer according to the centroid method. A single subtype predictor (SSP) applying the centroid method was used to classify 947 samples from six larger dataset into the molecular subtypes. For 787 of these samples followup information was available. Panels a–e show separate Kaplan–Meier analyses for the different molecular subtypes classified by the IL-8/B cell signature. A strong trend for significant difference was only observed in the Basal-like subtype (P = 0.061), while no prognostic value was detected in the remaining subtypes
Multivariate analysis of the prognostic value of the IL-8/B cell signature in TNBC
For 148 new TNBC samples that were not included in the finding cohort of our previous publication, both followup data and complete clinical information on lymph node status, tumor size, age, and histologic grading were available. Table 4 displays the results of a multivariate Cox regression including all these clinical parameters together with the IL-8/B cell signature. Only the IL-8/B cell signature was significant (HR 3.20, 95 % CI 1.43–7.16; P = 0.005) in that analysis.
Discussion
Triple negative breast cancers (TNBC) are clinically heterogeneous and prognostic markers, and biology-based therapies are needed to better treat this disease [4, 8]. By applying a subtype-specific approach, we have previously identified a gene signature which demonstrates significant prognostic value within this subgroup of breast cancers [31]. In our current study, we show that this signature assigns similar proportions of samples to “good” or “poor” prognosis even among other subtypes of breast cancer. However, we failed to detect any significant prognostic value of the signature in subtypes other than TNBC or basal-like breast cancer. These results demonstrate the importance of subtype-specific analyses both for the development and for the application of gene signatures in a clinical setting.
Regarding the cellular source of expression of the gene signature, we had already demonstrated that lymphocyte infiltration is responsible for the high level of expression of the B cell metagene [3, 30, 32], while IL-8 is expressed by the carcinoma cells themselves [31]. Supplementary Figure S1 displays the analyses of Affymetrix microarray data including microdissected samples which are in line with our previous results. So far, we could only demonstrate a pure prognostic value of our signature since better prognosis was observed both for patients treated with chemotherapy as well as those without adjuvant treatment [31]. We detected only a modest predictive value of the B cell metagene for response to neoadjuvant chemotherapy in our datasets [17, 31], despite that an independent larger study clearly demonstrated a predictive value of lymphocyte infiltration within the TNBC subgroup [6]. Several recent studies suggest that the IL-8/B cell gene signature could potentially provide predictive value for specific therapeutic approaches. Inhibition of IL-8 signaling has been suggested as a target to block breast cancer stem cells and cancer’s inflammatory roots [11, 20–22]. Consequentially, those TNBC patients characterized by high IL-8 expression and a poor prognosis would be first candidates for such an approach. On the other hand, T cell immunomodulators like anti-CTLA4 antibodies have shown great successes in some cancer patients at least in combination therapy [25, 33]. In this approach, it will be crucial to identify the right patients where one should take of the lid from immunosuppression [18, 26, 40] and a potential predictive value of the IL-8/B cell ratio could be worth testing.
Conclusions
A restriction of the prognostic value of different gene signatures and biologic processes to specific subtypes has been shown in many studies [2, 15, 23, 28, 29, 39]. For example, signatures consisting mainly of proliferation genes achieve their prognostic value only within ER-positive breast cancer. These subtype differences seem also to hold true for the next generation of genomic methods. Recent results from whole genome sequencing studies in breast cancer revealed that the number of somatic mutations varied markedly between individual tumors and the number of genes which were repeatedly found to be mutated at high frequency in is rather small [34, 37]. However, the frequency of mutations clearly differs between subtypes with TNBC and HER2-positive subtypes showing higher numbers of mutations than Luminal subtypes [1]. In addition, large scale integrated analyses of copy number variation and gene expression also suggest additional molecular stratification of breast cancer beyond the known intrinsic subtypes [5]. All these data underline the importance of stratified analyses in different subtypes and suggest a sequential multistep approach for application of future genomic tests in the clinic.
Abbreviations
- BLBC:
-
Basal-like breast cancer
- DMFS:
-
Distant metastasis free survival
- EFS:
-
Event free survival
- ER:
-
Estrogen receptor
- FNA:
-
Fine needle aspiration
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hazard ratio
- IL:
-
Interleukine
- PgR:
-
Progesterone receptor
- REMARK:
-
Recommendations for prognostic and tumor marker studies
- RFS:
-
Relapse free survival
- TNBC:
-
Triple negative breast cancer
References
Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Piña V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486(7403):405–409
Bianchini G, Iwamoto T, Qi Y, Coutant C, Shiang CY, Wang B, Santarpia L, Valero V, Hortobagyi GN, Symmans WF, Gianni L, Pusztai L (2010) Prognostic and therapeutic implications of distinct kinase expression patterns in different subtypes of breast cancer. Cancer Res 70(21):8852–8862
Bianchini G, Qi Y, Alvarez RH, Iwamoto T, Coutant C, Ibrahim NK, Valero V, Cristofanilli M, Green MC, Radvanyi L, Hatzis C, Hortobagyi GN, Andre F, Gianni L, Symmans WF, Pusztai L (2010) Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol 28(28):4316–4323
Carey L, Winer E, Viale G, Cameron D, Gianni L (2010) Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 7(12):683–692
Curtis C, Shah SP, Chin S, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Caldas C, Aparicio S, Brenton JD, Ellis I, Huntsman D, Pinder S, Purushotham A, Murphy L, Bardwell H, Ding Z, Jones L, Liu B, Papatheodorou I, Sammut SJ, Wishart G, Chia S, Gelmon K, Speers C, Watson P, Blamey R, Green A, Macmillan D, Rakha E, Gillett C, Grigoriadis A, Di Rinaldis E, Tutt A, Parisien M, Troup S, Chan D, Fielding C, Maia A, McGuire S, Osborne M, Sayalero SM, Spiteri I, Hadfield J, Bell L, Chow K, Gale N, Kovalik M, Ng Y, Prentice L, Tavaré S, Markowetz F, Langerød A, Provenzano E, Børresen-Dale A (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486(7403):346–352
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G (2010) Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28:105–113
Dupuy A, Simon RM (2007) Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst 99:147–157
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948
Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) Affy-analysis of affymetrix genechip data at the probe level. Bioinformatics 20(3):307–315
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10):R80
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan JL, Dontu G, Wicha MS (2010) CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest 120:485–497
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn H (2011) Strategies for subtypes-dealing with the diversity of breast cancer: highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer. Ann Oncol 22(8):1736–1747
Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, La Carey, Reynolds E, Dressler L, Nobel A, Parker J, Ewend MG, Sawyer LR, Wu J, Liu Y, Nanda R, Tretiakova M, Ruiz Orrico A, Dreher D, Palazzo JP, Perreard L, Nelson E, Mone M, Hansen H, Mullins M, Quackenbush JF, Ellis MJ, Olopade OI, Bernard PS (2006) The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 7:96
Hugh J, Hanson J, Cheang MCU, Nielsen TO, Perou CM, Dumontet C, Reed J, Krajewska M, Treilleux I, Rupin M, Magherini E, Mackey J, Martin M, Vogel C (2009) Breast cancer subtypes and response to docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. J Clin Oncol 27(8):1168–1176
Iwamoto T, Bianchini G, Booser D, Qi Y, Coutant C, Shiang CY, Santarpia L, Matsuoka J, Hortobagyi GN, Symmans WF, Holmes FA, O’Shaughnessy J, Hellerstedt B, Pippen J, Andre F, Simon R, Pusztai L (2011) Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J Natl Cancer Inst 103(3):264–272
Karn T, Metzler D, Ruckhäberle E, Hanker L, Gätje R, Solbach C, Ahr A, Schmidt M, Holtrich U, Kaufmann M, Rody A (2010) Data driven derivation of cutoffs from a pool of 3,030 Affymetrix arrays to stratify distinct clinical types of breast cancer. Breast Cancer Res Treat 120(3):567–579
Karn T, Pusztai L, Holtrich U, Iwamoto T, Shiang CY, Schmidt M, Müller V, Solbach C, Gaetje R, Hanker L, Ahr A, Liedtke C, Ruckhäberle E, Kaufmann M, Rody A, Ranganathan S (2011) Homogeneous datasets of triple negative breast cancers enable the identification of novel prognostic and predictive signatures. PLoS ONE 6(12):e28403
Karn T, Pusztai L, Ruckhäberle E, Liedtke C, Müller V, Schmidt M, Metzler D, Wang J, Coombes KR, Gätje R, Hanker L, Solbach C, Ahr A, Holtrich U, Rody A, Kaufmann M (2012) Melanoma antigen family A identified by the bimodality index defines a subset of triple negative breast cancers as candidates for immune response augmentation. Eur J Cancer 48(1):12–23
Kaufmann M, Pusztai L (2011) Use of standard markers and incorporation of molecular markers into breast cancer therapy: consensus recommendations from an international expert panel. Cancer 117(8):1575–1582
Korkaya H, Liu S, Wicha MS (2011) Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 121(10):3804–3809
Korkaya H, Liu S, Wicha MS (2011) Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots. Clin Cancer Res 17(19):6125–6129
Liu S, Wicha MS (2010) Targeting breast cancer stem cells. J Clin Oncol 28:4006–4012
Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, Gonzalez-Angulo AM, Pusztai L, Symmans WF, Bardelli A, Ellis P, Tutt ANJ, Gillett CE, Hennessy BT, Mills GB, Phillips WA, Piccart MJ, Speed TP, McArthur GA, Sotiriou C (2010) PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci USA 107(22):10208–10213
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics (2005) Reporting recommendations for tumor marker prognostic studies. J Clin Oncol 23:9067–9072
Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480(7378):480–489
Ogino S, Galon J, Fuchs CS, Dranoff G (2011) Cancer immunology—analysis of host and tumor factors for personalized medicine. Nat Rev Clin Oncol 8(12):711–719
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen La, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. Nature 406:747–752
Prat A, Ellis MJ, Perou CM (2011) Practical implications of gene-expression-based assays for breast oncologists. Nat Rev Clin Oncol 9(1):48–57
Reis-Filho JS, Pusztai L (2011) Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet 378(9805):1812–1823
Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, Solbach C, Hanker L, Ahr A, Metzler D, Engels K, Karn T, Kaufmann M (2009) T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res 11(2):R15
Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, Gaetje R, Solbach C, Ahr A, Metzler D, Schmidt M, Müller V, Holtrich U, Kaufmann M (2011) A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res 13(5):R97
Schmidt M, Hellwig B, Hammad S, Othman A, Lohr M, Chen Z, Boehm D, Gebhard S, Petry I, Lebrecht A, Cadenas C, Marchan R, Stewart JD, Solbach C, Holmberg L, Edlund K, Kultima HG, Rody A, Berglund A, Lambe M, Isaksson A, Botling J, Karn T, Müller V, Gerhold-Ay A, Cotarelo C, Sebastian M, Kronenwett R, Bojar H, Lehr H, Sahin U, Koelbl H, Gehrmann M, Micke P, Rahnenführer J, Hengstler JG (2012) A comprehensive analysis of human gene expression profiles identifies stromal immunoglobulin κ C as a compatible prognostic marker in human solid tumors. Clin Cancer Res 18(9):2695–2703
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331(6024):1565–1570
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SG, Morin GB, Watson P, Gelmon K, Chia S, Chin S, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486(7403):395–399
Simon RM, Paik S, Hayes DF (2009) Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst 101(21):1446–1452
Sotiriou C, Pusztai L (2009) Gene-expression signatures in breast cancer. N Engl J Med 360(8):790–800
Stephens PJ, Tarpey PS, Davies H, van Loo P, Greenman C, Wedge DC, Zainal SN, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Spencer Chapman M, Teague J, Easton D, Langerød A, OSBREAC, Lee MTM, Shen C, Tee BTK, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin S, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van ‘t Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SAJR, Salomon AV, Børresen-Dale A, Richardson A, Campbell PJ, Futreal PA, Stratton MR, Karesen R, Schlichting E, Naume B, Sauer T, Ottestad L (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486(7403):400–404
Weigelt B, Mackay A, A’Hern R, Natrajan R, Tan DSP, Dowsett M, Ashworth A, Reis-Filho JS (2010) Breast cancer molecular profiling with single sample predictors: a retrospective analysis. Lancet Oncol 11(4):339–349
Weigelt B, Pusztai L, Ashworth A, Reis-Filho JS (2011) Challenges translating breast cancer gene signatures into the clinic. Nat Rev Clin Oncol 9(1):58–64
Zitvogel L, Kepp O, Kroemer G (2011) Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 8(3):151–160
Acknowledgments
We thank Katherina Brinkmann and Samira Adel for expert technical assistance. This work was supported by grants from the the H.W. & J. Hector-Stiftung, Mannheim; the Margarete Bonifer-Stiftung, Bad Soden; and the BANSS-Stiftung, Biedenkopf.
Conflict of interest
The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Lars C. Hanker and Achim Rody contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10549_2012_2356_MOESM1_ESM.pdf
Supplementary Table S1: Datasets used in the study (n = 40) Description: A table containing detailed information on the datasets used in the study
10549_2012_2356_MOESM2_ESM.xls
Supplementary Table S2: Complete classification and assignment data for all 4,467 samples Description: An Excel table containing the complete classification and assignment data for each of the 4,467 samples and a respective link to the sample in public microarray databases
10549_2012_2356_MOESM3_ESM.pdf
Supplementary Table S3: Individual Affymetrix probesets of IL-8 metagene and B cell metagene Description: Two lists containing the Affymetrix probeset representing the IL-8 and B cell metagene, respectively
10549_2012_2356_MOESM4_ESM.pdf
Supplementary Figure S1: Expression of the B Cell and IL-8 metagenes in samples from core biopsies, fine needle aspiration, and microdissected tumor tissue Description: A figure demonstrating the expression of the B Cell and IL-8 metagenes in samples from core biopsies, fine needle aspiration, and microdissected tumor tissue
Rights and permissions
About this article
Cite this article
Hanker, L.C., Rody, A., Holtrich, U. et al. Prognostic evaluation of the B cell/IL-8 metagene in different intrinsic breast cancer subtypes. Breast Cancer Res Treat 137, 407–416 (2013). https://doi.org/10.1007/s10549-012-2356-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2356-2