
Abstract
The progression of ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) marks a critical step in the evolution of breast cancer. There is some evidence to suggest that dynamic interactions between the neoplastic cells and the tumour microenvironment play an important role. Using the whole-genome cDNA-mediated annealing, selection, extension and ligation assay (WG-DASL, Illumina), we performed gene expression profiling on 87 formalin-fixed paraffin-embedded (FFPE) samples from 17 patients consisting of matched IDC, DCIS and three types of stroma: IDC-S (<3 mm from IDC), DCIS-S (<3 mm from DCIS) and breast cancer associated-normal stroma (BC-NS; >10 mm from IDC or DCIS). Differential gene expression analysis was validated by quantitative real time-PCR, immunohistochemistry and immunofluorescence. The expression of several genes was down-regulated in stroma from cancer patients relative to normal stroma from reduction mammoplasties. In contrast, neoplastic epithelium underwent more gene expression changes during progression, including down regulation of SFRP1. In particular, we observed that molecules related to extracellular matrix (ECM) remodelling (e.g. COL11A1, COL5A2 and MMP13) were differentially expressed between DCIS and IDC. COL11A1 was overexpressed in IDC relative to DCIS and was expressed by both the epithelial and stromal compartments but was enriched in invading neoplastic epithelial cells. The contributions of both the epithelial and stromal compartments to the clinically important scenario of progression from DCIS to IDC. Gene expression profiles, we identified differential expression of genes related to ECM remodelling, and specifically the elevated expression of genes such as COL11A1, COL5A2 and MMP13 in epithelial cells of IDC. We propose that these expression changes could be involved in facilitating the transition from in situ disease to invasive cancer and may thus mark a critical point in disease development.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The multistep model of breast cancer progression is based on the morphological evolution from atypical ductal hyperplasia (ADH) to ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) [1–3]. Gene expression profiling-based studies have failed to demonstrate significant differences between these different stages of progression and instead have shown that multiple samples from an individual patient cluster closer to one another than to their respective stage of progression (ADH, DCIS and IDC) [4–7]. These, and other studies (combination of genomic, gene expression and immunohistochemical), suggest that the molecular phenotype is already established at the ADH/DCIS stage and does not change considerably as DCIS progresses to invasive cancer [2, 8–10]. Nevertheless, some subtle alterations have been identified and relate to processes such as epithelial mesenchymal transition, extracellular matrix (ECM) remodelling, and proliferation [5, 6, 11].
It is becoming increasingly apparent that the tumour microenvironment plays a key role in defining tumour behaviour and patient outcome [12]. Gene expression changes occur in cancer-associated stroma and are known to be implicated in prognosis as well as in cancer progression [5, 13–16]. Specifically, Ma et al. [5] provided strong evidence from gene expression profiling that the stroma co-evolves with the epithelial compartments during progression.
Gene expression profiling of breast cancer progression has so far only been studied using fresh frozen (FF) material [5] owing to the highest quality of RNA being available from this sample type. However, formalin-fixed paraffin-embedded tissue (FFPE) archives also provide a valuable resource for clinical research with wide availability of samples, particularly for different stages of progression. Gene expression profiling of FFPE clinical samples is also now feasible due to recent technological advances [17, 18]. Here we present the application of whole-genome cDNA-mediated annealing, selection, extension and ligation assay (WG-DASL, Illumina) [17, 18] to the study of the transition of DCIS to IDC in the context of both epithelial and stromal compartments.
Materials and methods
Tissue samples
FF and archival FFPE materials were accessed locally with approval from the Human Research Ethics Committee of the Royal Brisbane and Women’s Hospital (RBWH), Uniting HealthCare (The Wesley Hospital) and The University of Queensland. FFPE blocks from 17 patients specifically diagnosed with concurrent IDC and DCIS between 2007 and 2009 were selected for gene expression profiling (Table 1). All cases were pure invasive ductal carcinoma no special type (IDC NST) (n = 14) or mixed ductal lobular (n = 3; ductal component exclusively studied), of histological grade 2 or 3 [20, 21]. Only IDCs of a solid or glandular pattern with minimal intervening stroma were used. Three types of breast interlobular stroma were studied: IDC-S (stroma within 3 mm of IDC), DCIS-S (stroma within 3 mm of DCIS) and BC-NS (stroma > 10 mm from IDC and/or DCIS), Fig. 1. The BC-NS was obtained from either the same (n = 5) or an alternative paraffin block (n = 11) to the IDC/DCIS lesion. Additional cohorts of samples used, including reduction mammoplasties (RM, free from inflammation and fibrocystic change) are detailed in Online Resource Table S1.

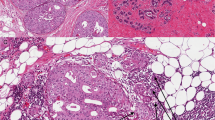
Epithelial and stromal components microdissected and analysed in this study. a Schematic detailing the two epithelial compartments (DCIS, IDC) and the three different cancer-associated stromal compartments selected (hatched). The respective distances from the epithelial lesions (DCIS or IDC) is given. b–d Haematoxylin and eosin stain of representative cases delineating regions microdissected: b focus of DCIS and associated stroma (within 3 mm; DCIS-S); c morphologically normal stroma dissected at a distance of >10 mm (BC-NS) from the lesion, in this instance, IDC; d focus of IDC and associated stroma (within 3 mm; IDC-S)
RNA extraction and quantitative real time-PCR (qRT-PCR)
Manual microdissection of epithelial (IDC and DCIS) and stromal compartments was performed, by a pathologist, using a stereomicroscope and a sterile needle. Up to 15 tissue sections for DCIS and IDC and 40–50 sections for the corresponding stromal samples were required. RNA was isolated using the High Pure RNA Paraffin kit (Roche Diagnostics Australia Pty Ltd., Castle Hill, NSW, Australia). cDNA synthesis was performed using 250 ng of total RNA and SuperScript™ III (Invitrogen Australia Pty Ltd., Mulgrave, VIC, Australia). TaqMan Gene Expression Assays (Applied Biosystems, Mulgrave, VIC, Australia; COL11A1, Hs00266273_m1; COL17A1, Hs00990073_m1; SFRP1, Hs00610060_m1; SOX10, Hs00366918_m1; MMP13, Hs00233992_m1; ABCB1, Hs01067802_m1 and MRAP2, Hs00536621_m1; USP19, Hs01103458_m1) were selected for qRT-PCR. Relative quantification using the comparative ΔΔCt method was performed [22], normalised to the endogenous control (RPL13A, Hs03043885_g1). A reference sample consisting of equal proportions of epithelial and stromal material was pooled from three RMs. The Mann–Whitney U statistical test was used to determine significance (P < 0.05).
Gene expression profiling and data analysis
Whole-genome DASL (Illumina, Scoresby, VIC, Australia) was performed as previously described [17]. Briefly, 200 ng of RNA from samples meeting quality criteria (sufficient RNA and a Ct value of < 29, as determined by qRT-PCR for RPL13A; RNA from all components of case 5 failed quality control and so the whole case was omitted from WG-DASL see Table 1) was processed using the MCS2 reagent and hybridised to the Human Ref8_V3_BeadChips. Data were collated using GenomeStudio (Illumina). The lumi [23] and limma [24] Bioconductor software packages using R (version 2.13.0) were used to perform quantile normalisation and determine differential expression respectively. Genes with Benjamini and Hochberg adjusted P values (< 0.05) with a greater than 50 % chance of being differentially expressed (positive B-statistic) were considered differentially expressed. Genespring GX 10.0.2 (Agilent Technologies, Mulgrave, VIC, Australia) was used for data visualisation and gene ontology (GO) analysis using P < 0.05. Normalised data can be accessed from GEO (GSE35019).
Immunohistochemistry (IHC) and immunofluorescence (IF) and analysis
IHC for SFRP1 (Abnova, Taipei City, Taiwan) and SOX10 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) was performed on FF and FFPE sections, respectively. The MACH 1 Universal HRP-Polymer kit (Biocare Medical, Concord, CA, USA) was used for detection. Further details are described in Online Resources. Percentage and intensity (weak = 1+, moderate = 2+, strong = 3+) of positive cytoplasmic and/or nuclear staining in normal and tumour epithelial cells were scored. Dual IF for COL11A1 and CK8/18 was performed, scored and analysed as described (Online Resource Methods and Figures S1 and S2).
Results
WG-DASL gene expression profiling
Of the 87 samples analysed by WG-DASL (Table 1), 69 passed average signal intensity (>250) and P95 (>800) criteria as described in [17]. Five IDC samples were performed in duplicate and showed an r 2 value between 0.89 and 0.97 (Online Resource Figure S3); 22,723 probes showed a reliable detection score (P < 0.01) in at least one sample. Unsupervised hierarchical clustering of probes which differed twofold from the mean (10,870 probes) showed that, apart from seven samples, there was a clear separation between stromal and epithelial samples (Fig. 2a). In total, 64 probes representing 58 genes were differentially expressed between IDC and DCIS (Table 2), of which 42 and 16 genes were up- and down-regulated in DCIS compared to IDC, respectively. Clustering of these 58 genes in the epithelial samples (DCIS and IDC) showed near complete segregation of the IDC and DCIS sample types (Fig. 2b; Online Resource Figure S4 details a K-means clustering analysis of this data).

WG-DASL expression analysis of the epithelial and stromal compartments of DCIS and IDC patient samples. a Hierarchical clustering of 69 successful samples using the 10,870 probes that demonstrated a two-fold change (up or down) across the sample set. Individual genes are arranged in rows and samples in columns. IDC and DCIS epithelial samples (black) tended to cluster separately from the stromal samples (white). b Supervised clustering analysis of the epithelial sample cohort using the 64 probes (58 genes) that were differentially expressed between DCIS and IDC (IDC grade 3, purple; IDC grade 2, pale blue; DCIS high-grade, yellow; DCIS intermediate (Int) grade, pink). c Unsupervised clustering of the stromal samples using all probes (>two fold from the mean across the dataset); IDC-S, red; DCIS-S, blue; BC-NS, maroon; RM-NS, grey)
Unsupervised clustering (Fig. 2c) separated normal stroma from healthy patients (RM-NS) and breast cancer patients (BC-NS) into the two major arms of the dendrogram. No obvious stratification was observed between the three types of stroma from breast cancer patients (BC-NS, IDC-S, DCIS-S) and no significantly differentially expressed genes were identified between any of these sample comparisons (i.e. IDC-S vs BC-NS; IDC-S vs DCIS-S; DCIS-S vs BC-NS). However, pairwise comparison of the cancer stromal types with the RM-NS samples showed that the same genes were consistently expressed at lower levels in the cancer stroma. Indeed, ten genes were differentially expressed between IDC-S and RM-NS and five of these were differentially expressed between DCIS and RM-NS (Table 2). Furthermore, two of these genes, USP19 and MRAP2, were significantly differentially expressed between the two most ‘normal’ stromal samples, RM-NS and BC-NS, while ABCB1 was down-regulated in all cancer-associated stromal samples compared to RM-NS.
Verification of microarray data by qRT-PCR
To validate the WG-DASL findings, qRT-PCR relative quantitation was performed for a selection of biologically interesting transcripts (COL17A1, COL11A1, SFRP1, SOX10, MMP13, ABCB1, USP19, MRAP2). MMP13, also known as collagenase-3, has been widely studied in breast cancer, where it has been shown that it can promote the DCIS to IDC transition [25, 26]. COL11A1 encodes a minor collagen found in many tissues; its expression has been shown to be dysregulated in cancers such as breast and colon [27–30]. COL17A1 is a transmembrane protein expressed in normal breast and involved in cell adhesion and SOX10, a transcription factor of the SRY family of the high mobility group box family, is known to be expressed in normal myoepithelial cells [31]. The SFRP1 gene, located at 8p11-21, is frequently altered or down-regulated in sporadic breast cancer and is implicated in cancer progression through its involvement with Wnt signalling [32]. The melanocortin-2 receptor accessory protein 2 (MRAP2) gene encodes a protein-coupled receptor protein, which regulates adrenocorticotropic hormone signalling [33], while USP19 is a de-ubiquitinating enzyme induced in skeletal muscle atrophy. ABCB1, also known as multidrug resistance gene 1 (MDR1), encodes a glycoprotein (PgP) that acts as an efflux pump, protecting the cells from xenobiotics [34] and has been implicated in breast cancer chemoresistance [35].
Three of five genes that were differentially expressed in DCIS versus IDC WG-DASL comparisons (SOX10, COL11A1 and MMP13) were confirmed as being differentially expressed by qRT-PCR (P < 0.05, Fig. 3a, b). There is considerable variation in expression levels between samples, as expected for human tumour samples. While WG-DASL and qRT-PCR data for COL17A1 expression was inconsistent, SFRP1 showed a consistent trend, and with removal of the two highest expressing IDC samples, significance was achieved (P ≤ 0.01). These two samples are the only ‘triple-negative’ tumours in the cohort (Table 1) and this phenotype is known to be associated with overexpression of SFRP1 (Online Resource Figure S5, [36]). COL11A1 was significantly up-regulated in IDC compared to both DCIS (P < 0.0001) and to its surrounding stroma (IDC-S; P = 0.012, Fig. 3b). IDC-S exhibited variable expression levels of COL11A1, but overall showed a significantly higher expression than the DCIS-S (P = 0.007). DCIS-S and BC-NS stromal samples, and those from reduction mammoplasties, showed consistently low levels of COL11A1 expression relative to IDC and IDC-S, with no significance observed in comparisons between the RM-NS and BC-NS samples (Fig. 3b).

Validation of gene expression changes in the epithelial and stromal components of DCIS and IDC lesions. Selected transcripts were validated using qRT-PCR; the data are normalised to RPL13A1, relative to a pooled normal reference and presented as an RQ (relative quantitation) value. Statistical significance was calculated using the Mann–Whitney U test (GraphPad Prism version 5) and is indicated only where significant (*P ≤ 0.03, **P ≤ 0.01). a Expression changes in the epithelial compartment of IDC and DCIS samples are presented for MMP13, SFRP1, SOX10 and COL17A1. b COL11A1 expression data for all sample types are presented. c Expression changes in ABCB1, MRAP and USP19 in the four stromal sample types (RM-NS, IDC-S, DCIS-S and BC-NS)
For the genes exclusively targeted in the stromal compartment, variable expression was detected. Two of four transcripts validated, again reflecting variability in human clinical samples; DCIS-S sample material was also limiting preventing strong correlations from being made (Fig. 3c). ABCB1 stromal transcripts showed concordance with the WG-DASL data, being significantly down-regulated in cancer stroma samples relative to the RM-NS (IDC-S, P < 0.001 and BC-NS, P < 0.05). MRAP2 qRT-PCR data did not replicate that of the WG-DASL, however, the cancer stroma shows a trend towards bimodality of expression, with divergent groups of high- and low- expressing samples. Exclusion of these ‘high-expressors’ in BC-NS and DCIS-S results in significantly lower levels of expression relative to the RM-NS, as observed by WG-DASL. USP19 expression was significantly different between the RM-NS and BC-NS samples (P = 0.008) yet contradicted the WG-DASL data, however, the presence of a single high expressing BC-NS sample likely skewed the statistics towards significance.
Verification of microarray data by IHC and IF
To validate expression changes at the protein level, we performed IHC for SFRP1 and SOX10, and IF for COL11A1 in independent cohorts of cases (Online Resource Table S2). SFRP1, evaluated in frozen tissue sections (Fig. 4a–c), was expressed in the cytoplasm of luminal cells of all normal TDLUs assessed (median percentage of cells stained 100 %; range 20–100 %). A significant decrease in expression was observed in DCIS (median 30 %; range 10–40 %; P = 0.004) and in IDC (median 20 %; range 0–60 % P = 0.0001) relative to the normal. There was no significant difference between DCIS and IDC. SOX10 was strongly expressed in normal myoepithelial cells of TDLUs and surrounding DCIS, however, expression was not detected in normal luminal epithelial cells, nor in 104/105 DCIS and invasive cancers (Fig. 4).

Immunohistochemical analysis of SFRP1 and SOX10 expression in NE, DCIS and IDC. a–c SFRP1 protein expression is observed in the normal luminal epithelial cells (arrowhead) as well as in stromal cells surrounding the TDLU; SFRP1 expression is reduced in DCIS (b) and in IDC (arrow in c) relative to NE (arrowhead in c). d–f SOX10 is a specific marker of myoepithelial cells of normal terminal ductal lobular units (arrowhead in d, f), and of DCIS (arrowhead in e), whereas luminal epithelial cells of normal TDLUs (d, f) and neoplastic epithelium of DCIS (e) and IDC (arrow in f) lack SOX10 expression
We performed IF staining on frozen sections to assess COL11A1 expression in the different breast compartments; epithelial localisation of COL11A1 was confirmed by co-staining with CK 8/18 (Online Resource Figure S2). High levels were observed in the normal epithelium (NE) from both breast cancer (BC-NE) and healthy patients (RM-NE; mean signal intensity, point analysis: 74.7 and 63.9, respectively). COL11A1 expression was higher in IDC compared to DCIS (mean signal intensity, point analysis; 73.9 versus 51.5, respectively), although this observation agreed with mRNA data, it was not significant, likely owing to limited sample size (Fig. 5). While COL11A1 was also expressed in both normal and neoplastic stroma, this expression was significantly lower compared to that of the epithelial compartments (BC-NE, DCIS and IDC, P ≤ 0.001). Fibroblasts were also shown to express COL11A1 (Online Resource Figure S6).

Dual immunofluorescence for COL11A1 and CK8/18 in breast cancer progression. Expression of COL11A1 was assessed in epithelial and stromal compartments of frozen sections from breast cancer patients containing normal TDLUs, DCIS and IDC and reduction mammoplasties containing normal TDLUs. Co-staining was performed with the epithelial marker CK8/18 (red) and COL11A1 (green) to highlight the presence of co-expression (see merged image—right hand panel). a–c Breast cancer case with normal TDLU (a), DCIS (b) and IDC (c) showing reduced expression of COL11A1 in DCIS relative to IDC. d, e Breast cancer case with DCIS (d) and IDC (e) where COL11A1 was expressed to similar levels between the epithelial compartments. f Reduction mammoplasty case showing high COL11A1 expression in epithelium of normal TDLU. The left hand panel shows the DAPI counterstain
Gene ontology analysis and meta-analysis
The list of genes differentially expressed between DCIS and IDC (n = 58) was subjected to GO analysis in an effort to identify the cellular pathways and processes that might be involved in the transition to invasive cancer (Online Resource Table S3). An interesting feature of this list was that 11/58 genes were associated with the ECM. In order to investigate whether stromal contamination was prompting this enrichment for ECM terms, we analysed a list of genes that were also differentially expressed between concurrent DCIS and IDC [6]. This study used FF material and laser capture microdissection (LCM) to obtain pure populations of neoplastic epithelial cells with minimal or no stromal contamination [6]. We attributed GO terms to the ‘Schuetz’ gene list [6] and also found an enrichment for functional terms related to ECM (Online Resource Table S3) in this exclusively epithelial analysis. Specifically, eight probes were shared between the Schuetz list and our own accounting for three genes: COL11A1, COL5A2 and MMP13. Comparing our differentially expressed gene list with another recent analysis of DCIS to IDC transition using a LCM-based approach [11], we found 11 genes to be shared. Intriguingly, of these 11 genes, there were six different collagens and three high molecular weight keratins (COL10A1, COL11A1, COL12A1, COL17A1, COL5A2, COL8A1, GPC6, KRT14, KRT17, KRT5, MYH11). We did not find any overlap between our stromal list and that of Knudsen et al. [11], nor with Hannemann et al. [37] who reported a 35-gene signature that can distinguish DCIS from IDC.
Discussion
We have applied gene expression profiling technology (WG-DASL, using MCS2 version reagents) to FFPE tissues to analyse the breast epithelial and stromal compartments in the context of tumour progression. Technically, the WG-DASL was successful, with five replicate pairs correlating well, ~80 % samples passing internal control criteria and epithelial and stromal samples stratifying as expected following unsupervised hierarchical clustering. Previous WG-DASL studies have focused on the technical feasibility of the assay, through for example, FF versus FFPE comparisons [18, 38, 39] and the validation of candidate genes (e.g. ER [40], Her2 [41]) related to subtyping of breast cancers. Nevertheless, gene expression profiling of archival FFPE samples remains extremely challenging, particularly for discovery approaches to understand important biological and/or clinical scenarios, as has been attempted here with respect to tumour progression. We were able to validate 5/9 differentially expressed genes using qRT-PCR and immunological techniques although we observed considerable variation in the expression levels of some transcripts across the clinical samples. It is difficult to conclude confidently whether this validation rate is appropriate, given the relatively limited number of published WG-DASL studies and that the starting material is FFPE, and therefore of highly variable quality. We would advocate validation of a larger panel of transcripts however, in this instance, clinical material was limiting.
The putative tumour suppressor, Secreted frizzled-related protein 1 (SFRP1) [42] was identified by WG-DASL as being down-regulated during progression from DCIS to IDC, consistent with other reports [5, 43, 44]. However this pattern of expression was validated by qRT-PCR only when the two high expressing IDC were removed from analysis. These ‘outliers’ were the only triple-negative tumours in the cohort studied and together with our meta-analysis of publically available microarray data (Online Resource Figure S4) confirms reports that loss of SFRP1 expression is associated with hormone receptor positivity [44] and conversely is a key phenotypic marker in some basal-like tumours [36]. Interestingly, the gene resides on chromosome 8p11-12 within the complex and variable amplification that is identified in ~10–15 % of breast cancers [45, 46]. Despite being part of this amplicon, SFRP1 gene expression is actually down-regulated due to gene methylation, leading to cancer progression through activation of Wnt signalling pathways [32]. The role of SFRP1 as a tumour suppressor in progression may therefore be restricted to certain tumour subtypes (luminal and HER2 related) and possibly a proportion of basal-like/triple-negative tumours.
Contrary to previous reports [4, 5, 7], we did not observe an enrichment of cell cycle-related genes differing between DCIS and IDC compartments. This observation, however, maybe a reflection of study design; the earlier studies used a mixture of low and high-grade tumours, whereas the current study used a more homogeneous cohort of mostly grade 3 tumours. It is perhaps as a consequence of this that we found just 58 differentially expressed genes between these epithelial components, highlighting the overall similarity between matched cases of DCIS and IDC. This is exemplified by the fact that a number of the genes ‘down-regulated’ in IDC compared to DCIS in this study are specific myoepithelial markers (e.g. SOX10, KRT14, KRT5, KRT17; Table 2) and as such were derived from only small population of DCIS-associated myoepithelial cells (Fig. 4). The high molecular weight cytokeratins are well established myoepithelial markers, whereas SOX10 was only recently described as a specific marker of normal myoepithelial cells [31]. The presence of SOX10 positive DCIS-associated myoepithelial cells, which were included in the microdissection of the DCIS samples, would account for the apparent SOX10 overexpression seen in DCIS relative to IDC by WG-DASL and qRT-PCR analyses.
Despite the lack of overlap in gene lists between the current study and previous reports [4, 5, 37], there remains some common gene families and biological processes that are featured in the transition of DCIS to IDC. For instance, in the current study there were a number of genes differentially expressed between DCIS and IDC epithelia that are related to the ECM (e.g. COL17A, COL5A2, COL22A1, COL8A1, COL12A1, COL10A1, COL11A1, MMP13, GPC6, KLK5, FREM1). Comparisons with reported studies [6, 11, 30, 47], including those that exclusively enriched for epithelial cells using LCM, supported our finding that the epithelial compartment is producing ECM-related components. Specifically, we showed by gene expression profiling, qRT-PCR and IF that COL11A1 is produced by the tumour epithelial cells to significantly higher levels than DCIS and the immediate adjacent stroma (IDC-S < 3 mm) suggesting that COL11A1, among other ECM proteins, might play a role in local invasion of breast cancer cells. In support of this, altered expression of COL11A1 has frequently been associated with tumour development and/or progression by others. Significant differential expression of this gene was recently reported to be the top hit in an array-based comparison of DCIS and IDC stroma and epithelium [11]. Turashvili et al. [47] found COL11A1 to be differentially up-regulated in invasive ductal and lobular carcinomas compared to NE (>six-fold change). Additionally, COL11A1 epithelial expression has also been demonstrated at the protein level, not only in breast tissue [29] but also in colorectal epithelium [27]. The co-expression and concomitant up-regulation of COL11A1 with COL5A2 have previously been shown in colorectal cancer compared to its precursor lesion [28, 29], which is in agreement with our data that showed up-regulation of both these genes in IDC relative to DCIS. Importantly, COL11A1 was found to be the top ranked gene in a late-stage ovarian, colorectal and breast cancer meta-analysis and is therefore a defining gene for a metastatic signature [48].
Gene expression changes in the cancer-associated stroma have proven important in predicting clinical outcome in breast cancer patients [13]. Other data indicates that the dynamic interaction between the epithelium and stroma plays an important role in governing the behaviour of a tumour [12]. Overall, we observed relatively few gene expression changes in the stromal samples along progression from normal through to IDC. This is consistent with recent reports where fewer gene expression changes were identified in the stromal compartments relative to the epithelial compartments during progression [11]. We exclusively used the interlobular stroma (surrounding the tumour, as opposed to intervening stroma) in order to avoid epithelial contamination within our stromal samples and this may explain the lack of correlation between our tumour–stroma gene lists and the two previously reported [5, 11]. This may also indicate that the majority of the stromal change occurs immediately adjacent to neoplastic epithelial cells where direct tumour–stroma interaction is most prominent. It must also be noted that these two published stromal signatures [5, 11] do not significantly overlap with each other, despite being performed with FF samples. We found a set of five genes (USP19, MRAP2, EFCBP1, ABCB1 and FADH1) to be differentially expressed only when compared with normal stromal samples from reduction mammoplasties (RM-NS) using WG-DASL. Further, the most ‘normal’ stromal samples (BC-NS and RM-NS) did not cluster together in the hierarchical clustering, suggesting that subtle gene expression changes exist between normal stroma from healthy and cancer patients.
In summary, we have applied the WG-DASL assay for exploring gene expression pattern changes to address the clinically important scenario of progression from DCIS to IDC and the role played by both the epithelial and stromal compartments in this process. We found that the majority of expression changes during progression occurred within the epithelial cell compartments with relatively little change occurring in the stroma. Consistent with previous reports was the enrichment in biological processes related to ECM remodelling in IDC compared to DCIS, and specifically this related to the elevated expression of genes such as COL11A1, COL5A2 and MMP13 in epithelial cells of IDC. These genes might therefore be playing a crucial role in facilitating the invasion of neoplastic cells into and through the surrounding stroma.
Abbreviations
- BC-NS:
-
Breast cancer normal stroma
- DCIS:
-
Ductal carcinoma in situ
- DCIS-S:
-
DCIS-stroma
- ECM:
-
Extracellular matrix
- FF:
-
Fresh frozen
- FFPE:
-
Formalin fixed paraffin embedded
- IDC:
-
Invasive ductal carcinoma
- IDC-S:
-
IDC-stroma
- IF:
-
Immunofluorescence
- IHC:
-
Immunohistochemistry
- ILC:
-
Invasive lobular carcinoma
- LCM:
-
Laser capture microdissection
- NE:
-
Normal epithelium
- qRT-PCR:
-
Quantitative real time-PCR
- RM:
-
Reduction mammoplasty
- RM-NS:
-
Reduction mammoplasty normal stroma
- WG-DASL:
-
Whole-genome cDNA-mediated annealing, selection, extension and ligation assay
References
Wellings SR, Jensen HM (1973) On the origin and progression of ductal carcinoma in the human breast. J Natl Cancer Inst 50(5):1111–1118
Simpson PT, Reis-Filho JS, Gale T, Lakhani SR (2005) Molecular evolution of breast cancer. J Pathol 205(2):248–254
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M, Marchio C, Reis-Filho JS (2010) Breast cancer precursors revisited: molecular features and progression pathways. Histopathology 57(2):171–192
Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC (2003) Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA 100(10):5974–5979
Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC (2009) Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 11(1):R7
Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D, Kurek R, Neubauer HJ (2006) Progression-specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res 66(10):5278–5286
Castro NP, Osorio CA, Torres C, Bastos EP, Mourao-Neto M, Soares FA, Brentani HP, Carraro DM (2008) Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma. Breast Cancer Res 10(5):R87
Gao Y, Niu Y, Wang X, Wei L, Lu S (2009) Genetic changes at specific stages of breast cancer progression detected by comparative genomic hybridization. J Mol Med 87(2):145–152
Lakhani SR, Collins N, Stratton MR, Sloane JP (1995) Atypical ductal hyperplasia of the breast: clonal proliferation with loss of heterozygosity on chromosomes 16q and 17p. J Clin Pathol 48(7):611–615
Heaphy CM, Bisoffi M, Joste NE, Baumgartner KB, Baumgartner RN, Griffith JK (2009) Genomic instability demonstrates similarity between DCIS and invasive carcinomas. Breast Cancer Res Treat 117:17–24
Knudsen ES, Ertel A, Davicioni E, Kline J, Schwartz GF, Witkiewicz AK (2011) Progression of ductal carcinoma in situ to invasive breast cancer is associated with gene expression programs of EMT and myoepithelia. Breast Cancer Res Treat [Epub ahead of print]
Beck AH, Sangoi AR, Leung S, Marinelli RJ, Nielsen TO, van de Vijver MJ, West RB, van de Rijn M, Koller D (2011) Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci Transl Med 3(108):108–113
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14(5):518–527
West RB, Nuyten DS, Subramanian S, Nielsen TO, Corless CL, Rubin BP, Montgomery K, Zhu S, Patel R, Hernandez-Boussard T, Goldblum JR, Brown PO, van de Vijver M, van de Rijn M (2005) Determination of stromal signatures in breast carcinoma. PLoS Biol 3(6):e187
Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO (2004) Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2(2):E7
Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, Andre S, Piccart M, Campone M, Brain E, Macgrogan G, Petit T, Jassem J, Bibeau F, Blot E, Bogaerts J, Aguet M, Bergh J, Iggo R, Delorenzi M (2009) A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med 15(1):68–74
Waddell N, Cocciardi S, Johnson J, Healey S, Marsh A, Riley J, da Silva L, Vargas AC, Reid L, Simpson PT, Lakhani SR, Chenevix-Trench G (2010) Gene expression profiling of formalin-fixed, paraffin-embedded familial breast tumours using the whole genome-DASL assay. J Pathol 221(4):452–461
April C, Klotzle B, Royce T, Wickham-Garcia E, Boyaniwsky T, Izzo J, Cox D, Jones W, Rubio R, Holton K, Matulonis U, Quackenbush J, Fan JB (2009) Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples. PLoS ONE 4(12):e8162
Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A (eds) (2010) AJCC cancer staging manual, 7th edn. Springer, New York
Tavassoli FA, Devilee P (2003) World Health Organization Classification of Tumours. Pathology and genetics of tumours of the breast and female genital organs. IARC Press, Lyon
Elston CW, Ellis IO (1991) Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19(5):403–410
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408
Du P, Kibbe WA, Lin SM (2008) Lumi: a pipeline for processing Illumina microarray. Bioinformatics 24(13):1547–1548
Smyth GK, Michaud J, Scott HS (2005) Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21(9):2067–2075
Rizki A, Weaver VM, Lee SY, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR, Mian IS, Borowsky AD, Jensen RA, Idowu MO, Chen F, Chen DJ, Petersen OW, Gray JW, Bissell MJ (2008) A human breast cell model of preinvasive to invasive transition. Cancer Res 68(5):1378–1387
Delassus GS, Cho H, Eliceiri GL (2011) New signaling pathways from cancer progression modulators to mRNA expression of matrix metalloproteinases in breast cancer cells. J Cell Physiol 226(12):3378–3384
Bowen KB, Reimers AP, Luman S, Kronz JD, Fyffe WE, Oxford JT (2008) Immunohistochemical localization of collagen type XI alpha1 and alpha2 chains in human colon tissue. J Histochem Cytochem 56(3):275–283
Fischer H, Salahshor S, Stenling R, Bjork J, Lindmark G, Iselius L, Rubio C, Lindblom A (2001) COL11A1 in FAP polyps and in sporadic colorectal tumors. BMC Cancer 1:17
Fischer H, Stenling R, Rubio C, Lindblom A (2001) Colorectal carcinogenesis is associated with stromal expression of COL11A1 and COL5A2. Carcinogenesis 22(6):875–878
Halsted KC, Bowen KB, Bond L, Luman SE, Jorcyk CL, Fyffe WE, Kronz JD, Oxford JT (2008) Collagen alpha1(XI) in normal and malignant breast tissue. Mod Pathol 21(10):1246–1254
Nonaka D, Chiriboga L, Rubin BP (2008) Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol 32(9):1291–1298
Yang ZQ, Liu G, Bollig-Fischer A, Haddad R, Tarca AL, Ethier SP (2009) Methylation-associated silencing of SFRP1 with an 8p11-12 amplification inhibits canonical and non-canonical WNT pathways in breast cancers. Int J Cancer 125(7):1613–1621
Webb TR, Clark AJ (2010) Minireview: the melanocortin 2 receptor accessory proteins. Mol Endocrinol 24(3):475–484
Saeki T, Tsuruo T, Sato W, Nishikawsa K (2005) Drug resistance in chemotherapy for breast cancer. Cancer Chemother Pharmacol 56(Suppl 1):84–89
Krech T, Scheuerer E, Geffers R, Kreipe H, Lehmann U, Christgen M (2012) ABCB1/MDR1 contributes to the anticancer drug-resistant phenotype of IPH-926 human lobular breast cancer cells. Cancer Lett 315(2):153–160
Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, Gaetje R, Solbach C, Ahr A, Metzler D, Schmidt M, Muller V, Holtrich U, Kaufmann M (2011) A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res 13(5):R97
Hannemann J, Velds A, Halfwerk JB, Kreike B, Peterse JL, van de Vijver MJ (2006) Classification of ductal carcinoma in situ by gene expression profiling. Breast Cancer Res 8(5):R61
Kibriya MG, Jasmine F, Roy S, Paul-Brutus RM, Argos M, Ahsan H (2010) Analyses and interpretation of whole-genome gene expression from formalin-fixed paraffin-embedded tissue: an illustration with breast cancer tissues. BMC Genomics 11:622
Mittempergher L, de Ronde JJ, Nieuwland M, Kerkhoven RM, Simon I, Rutgers EJ, Wessels LF, Van’t Veer LJ (2011) Gene expression profiles from formalin fixed paraffin embedded breast cancer tissue are largely comparable to fresh frozen matched tissue. PLoS ONE 6(2):e17163
Ton CC, Vartanian N, Chai X, Lin MG, Yuan X, Malone KE, Li CI, Dawson A, Sather C, Delrow J, Hsu L, Porter PL (2011) Gene expression array testing of FFPE archival breast tumor samples: an optimized protocol for WG-DASL sample preparation. Breast Cancer Res Treat 125(3):879–883
Reinholz MM, Eckel-Passow JE, Anderson SK, Asmann YW, Zschunke MA, Oberg AL, McCullough AE, Dueck AC, Chen B, April CS, Wickham-Garcia E, Jenkins RB, Cunningham JM, Jen J, Perez EA, Fan JB, Lingle WL (2010) Expression profiling of formalin-fixed paraffin-embedded primary breast tumors using cancer-specific and whole genome gene panels on the DASL(R) platform. BMC Med Genom 3:60
Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck J, Knuchel R, Klopocki E, Sauter G, Simon R, Wieland WF, Walter B, Denzinger S, Hartmann A, Hammerschmied CG (2007) Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene 26(38):5680–5691
Suzuki H, Toyota M, Carraway H, Gabrielson E, Ohmura T, Fujikane T, Nishikawa N, Sogabe Y, Nojima M, Sonoda T, Mori M, Hirata K, Imai K, Shinomura Y, Baylin SB, Tokino T (2008) Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer 98(6):1147–1156
Ugolini F, Charafe-Jauffret E, Bardou VJ, Geneix J, Adelaide J, Labat-Moleur F, Penault-Llorca F, Longy M, Jacquemier J, Birnbaum D, Pebusque MJ (2001) WNT pathway and mammary carcinogenesis: loss of expression of candidate tumor suppressor gene SFRP1 in most invasive carcinomas except of the medullary type. Oncogene 20(41):5810–5817
Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, Lasorsa L, Letessier A, Ginestier C, Monville F, Esteyries S, Adelaide J, Esterni B, Henry C, Ethier SP, Bibeau F, Mozziconacci MJ, Charafe-Jauffret E, Jacquemier J, Bertucci F, Birnbaum D, Theillet C, Chaffanet M (2005) Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res 3(12):655–667
Courjal F, Cuny M, Simony-Lafontaine J, Louason G, Speiser P, Zeillinger R, Rodriguez C, Theillet C (1997) Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res 57(19):4360–4367
Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J, Hajduch M, Murray P, Kolar Z (2007) Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 7:55
Kim H, Watkinson J, Varadan V, Anastassiou D (2010) Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC Med Genom 3:51
Acknowledgments
We thank all the patients and their families for the donation of samples for research. We thank Rebecca Johnston and Julie Johnson for excellent technical assistance. Ana Cristina Vargas was supported by a clinical fellowship from the Ludwig Institute for Cancer Research (LICR); Peter T Simpson is the recipient of a Fellowship from the National Breast Cancer Foundation, Australia. Georgia Chenevix-Trench is a Senior Principal Research Fellow of the National Health and Medical Research Council (NHMRC) of Australia. This work was funded in part by grants from the LICR, the NHMRC, the Australian Medical Association Queensland and the Queensland Health Pathology Service—Study, Education and Research Trust Fund.
Conflict of interest
PTS received funding from Illumina to present at a conference about the WG-DASL assay. The remaining authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ana Cristina Vargas and Amy E McCart Reed, and Peter T Simpson and Sunil R Lakhani contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Vargas, A.C., Reed, A.E.M., Waddell, N. et al. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast Cancer Res Treat 135, 153–165 (2012). https://doi.org/10.1007/s10549-012-2123-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2123-4