
Abstract
Women with triple-negative breast cancer (TNBC) do not benefit from endocrine therapy or trastuzumab. Chemotherapy is the only systemic therapy currently available. To reduce the elevated risk of disease progression in these patients, better treatment options are needed, which are less toxic and more targeted to this patient population. We performed a comprehensive analysis of potential targetable genetic aberrations affecting the receptor tyrosine kinase/RAS/MAPK pathway, which are observed at higher frequencies in adenocarcinomas of other organs. Sixty-five individual TNBCs were studied by sequence analysis for HER2 (exon 18–23), EGFR (exon 18–21), KRAS (exon 2), and BRAF (exon 15) mutations. In addition, a tissue microarray was constructed to screen for EGFR gene copy gain and EML4-ALK fusion by FISH. Triple-negative status was confirmed by immunohistochemistry and FISH on tissue microarray sections. EGFR and CK5/6 immunohistochemical analyses were performed for identification of the basal-like phenotype. In addition, mutation analysis of TP53 (exon 5–8) was included. Sequence analysis revealed HER2 gene mutation in only one patient (heterozygous missense mutation in exon 19: p.L755S). No mutations were found in EGFR, KRAS, and BRAF. High polysomy of EGFR was detected in 5 of the 62 informative cases by FISH. True EGFR gene amplification accompanied by strong membranous EGFR protein expression was observed in only one case. No rearrangement of the ALK gene was detected. Basal-like phenotype was identified in 38 of the 65 TNBCs (58.5 %). TP53 gene mutation was found in 36/63 (57.1 %) tumors. We conclude that targetable genetic aberrations in the receptor tyrosine kinase/RAS/MAPK pathway occur rarely in TNBC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Triple-negative breast cancer (TNBC), defined by the lack of estrogen receptor (ER) and progesterone receptor (PR) expression and the absence of HER2/neu (ERBB2) gene amplification, accounts for approximately 15 % of all breast carcinomas [1]. Although TNBC is common in patients with BRCA1 mutations, it is not restricted to this group [2]. TNBCs represent a heterogeneous group of tumors in terms of presentation, morphology, and molecular aberrations. These tumors may present as invasive ductal (not otherwise specified, NOS), metaplastic, medullary, apocrine, or other histological type. However, most of them share distinct phenotypic and genotypic features like high tumor grade and accelerated tumor cell proliferation [1]. Molecular profiling has shown a close association to the basal-like subtype [3]. However, there are TNBCs that belong to other intrinsic subtypes, i.e., the claudin-low and molecular apocrine type [4]. TNBC has attracted the attention of both pathologists and oncologists as an “easily recognizable” prognostic group. Triple-negative tumors are associated with shorter time to recurrence and higher mortality, and the affected women are younger compared to the total group of breast cancer patients [5, 6]. Currently, TNBCs lack the benefit of available targeted therapy. Hormone therapies and HER2-targeted therapies are ineffective in their treatment, and thus, searching for new drug targets selective for this subtype of tumors is a major challenge in modern oncology [7].
Activation of distinct growth factor receptor pathways by mutation and/or amplification of the corresponding genes are a major mechanism in the development and progression of cancer and a main target for newly developed drugs. Up to 20 % of breast cancers show amplification of HER2 which is a strong predictive marker for therapy with monoclonal antibodies directed against HER2 (trastuzumab) [8]. HER2 belongs to the family of receptor tyrosine kinases. This family includes additional targetable proto-oncogenes such as the epidermal growth factor (EGFR) and the anaplastic lymphoma kinase (ALK). In adenocarcinomas of the lung, EGFR mutation/amplification and EML4-ALK fusion is frequently found, but HER2 amplification is less common [9]. The introduction of tyrosine kinase inhibitors against EGFR and ALK expanded the therapy options for lung cancer. In addition, activating mutations of HER2 and BRAF have been described at low frequency in adenocarcinoma of the lung and might be targeted by inhibitory drugs in the future [9]. As mutations of all these proto-oncogenes, along with KRAS, are involved in the activation of the same transforming MAP-kinase pathway, their occurrence is mutually exclusive [10].
In breast cancer, genetic aberrations of these biomarkers are described with low frequencies. EGFR mutations are reported in 0–11 % of breast cancer [11, 12], while EGFR amplifications are found in 1–6 % [13–16]. Two studies on EML4-ALK fusion report a frequency of 0 and 2 % [17, 18]. Mutations of the HER2 gene in breast cancer were found in 4/94 (4.3 %) tumors in one study [11]. KRAS mutations are found in 5 % of breast cancer [19], while BRAF mutations have been identified only in breast cancer cell lines and three primary tumors so far [20–22]. However, breast cancer is a heterogeneous group of diseases with divergent molecular mechanisms of pathogenesis. We hypothesized that mutations of these targetable oncogenic kinases, which are observed at higher frequencies in adenocarcinomas of other organs that are also not endocrine responsive, might accumulate in the group of hormone receptor-negative breast cancers without activation of the MAP-kinase pathway by HER2 amplification. We therefore performed a comprehensive analysis of potential targetable mutations and amplifications affecting the receptor tyrosine kinase/RAS/MAPK pathway, including EGFR mutation, EGFR amplification, HER2 mutation, EML4-ALK fusion, KRAS mutation, and BRAF mutation, in 65 triple-negative primary breast cancers. In addition, we performed TP53 mutation analysis in all cases.
Materials and methods
Specimen collection and TMA construction
All TNBCs diagnosed at the Institute of Pathology, University Medical Center Hamburg-Eppendorf, between January 2008 and January 2011 were reviewed for this study. All 65 cases with available surgical specimens of the primary tumor and a tumor size larger than 5 mm were included (one patient with a tumor size of 4 mm and one patient with complete remission after neoadjuvant chemotherapy were excluded). Negativities for hormone receptors (ER and PR) and HER2 were assessed during initial diagnostic procedures by immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH), respectively, on core biopsy or surgical specimen following the same procedures as described later. Five of the patients had received neoadjuvant chemotherapy, including taxanes, anthracyclines, cyclophosphamide, and 5-fluorouracil, before surgery. All cases were reviewed for histological type and grade. The pathologic stage was obtained from the original pathology report.
A tissue microarray (TMA) of the 65 TNBC specimens was constructed as previously described [23]. Each case is represented in duplicate on the TMA. Consecutive sections of the TMA were used for H&E-stained reference, immunohistochemical analysis, and FISH.
Immunohistochemistry
Immunohistochemical analyses were performed on 4-μm-thick TMA sections. Staining for ER alpha (ER; Clone 6F11, Novocastra, dilution scale 1:80), progesterone receptor (PR; Clone 16, Novocastra, dilution scale 1:200), and cytokeratin 5/6 (Clone D5/16B4, Dako, dilution scale 1:100) was carried out after heat pretreatment in Bond Epitope Retrieval Solution 2 (pH9; Leica Microsystems) in a Bond automated system (Leica Microsystems). HER2 (HercepTest, Dako) and EGFR (EGFR pharmDx, Dako) IHC were performed in an Autostainer (DAKO). The test kits were applied exactly according to the manufacturer’s instructions. ER and PR stains were considered negative if immunostaining was seen in <1 % of tumor nuclei. EGFR and CK5/6 stains were considered positive if any (weak, moderate, or strong) cytoplasmic and/or membranous stainings were observed [24]. For HER2 status, tumors were considered negative if scored 0 or 1+ according to the HercepTest criteria. FISH ratio was used to segregate immunohistochemical equivocal (2+) results (see below).
DNA extraction and targeted sequence analysis
DNA was extracted from formalin-fixed and paraffin-embedded tissues. A representative area containing at least 60 % tumor cells was marked on H&E sections of the original tumor blocks and sampled by punching two tissue cores with a diameter of 0.6 mm. DNA was extracted as per standard protocols (QIAmp DNA Mini Kit, Qiagen). Quantity and quality of DNA were evaluated using a Nanodrop spectrophotometer ND-1000 (Thermo-Scientific). 100 ng of DNA was subjected to PCR using the AmpliTaq Gold PCR master mix (Applied Biosystems) under the conditions recommended by the manufacturer. Primer sequences for HER2 (Exons 18–23), KRAS (Exon 2), and BRAF (Exon 15) are given in supplementary Table 1. PCR and primer sequences for EGFR (Exons 18–21) and TP53 (Exons 5–8) have been previously described [25, 26]. After verification of the amplicons on QIAxcel system (Qiagen), PCR products were purified using an enzymatic method (ExoSAP-IT; USB Products) and subjected to sequencing reactions using BigDye Terminator Cycle v1.1 Sequencing Kit (Applied Biosystems). Sequence reactions were analyzed on ABI PRISM 3100 genetic analyzer (Applied Biosystems).
FISH
FISH was performed on 4-μm-thick TMA sections using a commercially available kit for proteolytic slide pretreatment (paraffin pretreatment reagent kit, Abbott Molecular). HER2 FISH was carried out using the PathVysion kit (Abbott Molecular). EGFR copy gain was assessed using the Vysis EGFR/CEP7 FISH Probe Kit (Abbott Molecular). Vysis LSI ALK Dual Color, Break Apart Rearrangement Probe (Abbott Molecular) was used to detect any rearrangement involving the ALK gene. FISH preparation was carried out exactly according to the manufacturer’s guidelines. HER2 status was considered negative (not amplified) if the ratio of gene signal to centromere signal number was below 2. EGFR FISH was evaluated according to the suggested guidelines for non-small cell lung cancer by the Colorado group [27, 28]. Tumors were classified as EGFR FISH-positive if EGFR amplification or high polysomy was detected. Diagnosis of ALK rearrangement required rearranged FISH signals in more than 15 % of tumor cells as suggested by the manufacturer for the evaluation of non-small cell lung cancer.
Results
Study group
Triple-negative primary breast cancers of 65 patients were included in the study. All patients were women with a mean age of 56.1 years (range 32–86) at the time of surgery. Triple-negative status of all included tumors was confirmed by ER, PR, and HER2 IHC, as well as HER2 FISH on TMA sections representing two samples of each tumor. Basal-like phenotype was identified in 38 of the 65 analyzed TNBCs (58.5 %) using the five biomarker surrogate that includes cytokeratin 5/6 and EGFR IHC [24]. An overview of the studied cases is given in Table 1.
Mutation analysis of EGFR, HER2, KRAS, BRAF, and TP53
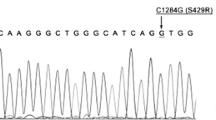
Sequence analysis of all exons was successful in 63 tumors. In two samples, severe DNA degradation after formalin fixation impeded successful amplification in some reactions. No mutations were found in the tyrosine kinase domain (exons 18–21) of EGFR (0/64). HER2 sequence analysis of exons 18–23 revealed mutation in one case (1/64). This tumor of a premenopausal patient showed high-grade morphology of ductal type (G3, pT1c, pN1). A heterozygous missense mutation in exon 19 of the HER2 gene, p.L755S (c.2264T>C), was detected (Fig. 1). The mutation was confirmed to be of somatic nature, and the tumor of an axillary lymph-node metastasis also showed this mutation. Using laser-capture microdissection, the mutation was also confirmed in the concomitant intraductal tumor component (DCIS). HER2 expression as assessed by IHC was negative (1+), and an additional mutation of the TP53 gene (p.A138S) was detected in this tumor. No mutations were found in KRAS, codon G12 and G13 (0/63), and BRAF, codon V600 (0/63). We found TP53 mutations within exons 5–8 in 36/63 (57.1 %) tumors. These mutations comprised 21 missense mutations, five nonsense mutations, nine frameshift mutations, and one in-frame deletion. This TP53 mutation frequency is in line with previously published results in TNBC [29] and provides evidence that the applied method of mutation analysis by Sanger sequencing of enriched tumor cells is of sufficient sensitivity.

HER2 mutation in case 61. Sequence analysis of primary invasive carcinoma (a), lymph-node metastasis (b), and carcinoma in situ (c) indicates substitution c.2264T>C (p.L755S) of the HER2 gene. Non-neoplastic tissue shows wild-type sequence (d). H&E stained detail of the primary tumor is shown in (e)
FISH analysis of EGFR gene copy gain and ALK rearrangement
FISH analysis on the TMA was successful in 62/65 cases for EGFR and 57/65 cases for ALK. The two cases, which failed to be amplified due to low DNA quality, also failed in both FISH assays. EGFR FISH showed high polysomy in five cases, i.e., ≥4 EGFR copies in ≥40 % of cells [28]. Two of these cases showed moderate expression of the EGFR protein, while in the other three cases, no or weak expression was detectable by IHC. True EGFR amplification with 30–50 gene copies in clusters accompanied by strong membranous EGFR protein expression of the tumor cells was found in one case (Fig. 2). This premenopausal patient was treated with neoadjuvant chemotherapy (CAT regime) before surgery, which did not result in complete remission. EGFR amplification was also detected in axillary and supraclavicular lymph-node metastases removed before neoadjuvant treatment indicating that the therapy regimen had no influence on the development of the EGFR amplification. The tumor showed high-grade morphology of ductal type (G3, ypT1b, pN3), and a concomitant TP53 mutation (p.R273C) was detected. No rearrangement of the ALK gene indicating EML4-ALK fusion was detected in any tumor.

EGFR amplification in case 64. EGFR FISH analysis (a) shows 30–50 EGFR gene copies per tumor cell nucleus in clusters (red signals) and 2 CEP7 signals (green). EGFR IHC; (b) demonstrates strong membranous EGFR expression. H&E stained detail of the primary tumor is shown in (c)
Discussion
Triple-negative breast cancer represents a major clinical challenge due to its aggressive behavior that comes along with a lack of available therapy targets. Until now, it is unknown what genetic aberrations induce self-sufficiency in growth signals in these tumors that are endocrine non-responsive and do not exhibit HER2 gene amplification. HER2 is a receptor tyrosine kinase and gene amplification results in an activation of the RAS/MAPK pathway. Studies of hormone receptor-negative adenocarcinomas of other organs have shown the importance of the receptor tyrosine kinase/RAS/MAPK pathway for tumor growth even in the absence of HER2 gene amplification. Driver mutations affecting this pathway have been well documented in non-small cell lung cancer. These genetic aberrations with potential clinical relevance for targeted therapy include EGFR mutation and amplification, EML4-ALK fusion, HER2 mutation, KRAS mutation, and BRAF mutation. In breast cancer, these mutations have been only punctually studied and are observed at low frequencies. Since these studies included hormone receptor positive and HER2 amplified breast cancers, we hypothesized that these infrequent driver mutations might accumulate in the group of TNBCs.
To the best of our knowledge, this study represents the first comprehensive and concurrent analysis of these potentially targetable driver mutations in a cohort of triple-negative breast cancers. We analyzed these tumors for activating mutations of EGFR, HER2, BRAF, and KRAS. In addition, a screening for ALK rearrangement and EGFR gene copy gain was performed by FISH. We found genetic aberrations in only two of the examined cases. One tumor harbors a HER2 missense mutation p.L755S. The mutation is also detectable in the in situ component (DCIS) of the tumor, suggesting an early event in the carcinogenesis of this tumor. This type of mutation affects the kinase domain of HER2 and has been previously described in four tumors including two breast cancers [11]. As shown in lung cancer, HER2 inhibitors such as lapatinib or afatinib might well be effective in tumors with activating HER2 mutations [30, 31]. The second case shows high level EGFR gene amplification which might render the tumor amenable to either EGFR tyrosine kinase inhibitors or monoclonal antibodies directed against EGFR as described for EGFR-amplified lung cancers [32–34].
Only few data are available on these predictive markers in TNBC. Gumuskaya et al. [15] reported EGFR gene amplification in one and high polysomy in 9 of 62 TNBCs, which is consistent with our data. In contrast to our results, Teng et al. have reported EGFR mutations in 8/70 TNBCs in their study performed in Singapore. We did not detect any EGFR mutations. Remarkably, EGFR mutations in lung cancer are much more frequent in Asian than in the Caucasian populations [35]. One might speculate that the frequency of EGFR mutations in breast cancer might also be higher in Asian populations. However, two studies from Japan and Korea did not find any EGFR mutations in a total of 177 unselected breast cancers [36, 37]. Therefore, it is currently unclear if the frequency of EGFR mutations in TNBC depends on the analyzed population. In accordance with our results, a recently published study on KRAS mutations in TNBCs found no mutations in 35 tumors [38]. Because of the small sizes of cohorts published so far, a systematic review of these studies is needed to accurately analyze the frequency of the different mutations and association with distinct populations.
Molecular profiling has shown that TNBC and basal-like subtype are closely associated but not congruent. To ensure the representativeness of our study collective, we used a five-biomarker surrogate immunopanel to identify the rate of basal-like phenotype in our collective. Consistent with the literature, 58.5 % of the analyzed TNBC were basal using the five-marker method [24, 39].
In summary, we found that the analyzed targetable genetic aberrations in the receptor tyrosine kinase/RAS/MAPK pathway are rare in TNBC. Therefore, these potential predictive markers might be helpful only in individual patients with TNBC. Our data support the view that TNBC is a biologically peculiar group of breast cancers. As these tumors only rarely harbor activating aberrations of tyrosine kinase receptors, their mechanism of independency on growth factors is still unclear and differs from the already known pathways in breast cancer.
References
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948
Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N, Trudel M, Akslen LA (2003) Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 95(19):1482–1485
Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, Gilks CB, van de Rijn M, Perou CM (2004) Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res 10(16):5367–5374
Perou CM (2011) Molecular stratification of triple-negative breast cancers. Oncologist 16(Suppl 1):61–70
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15 Pt 1):4429–4434
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V (2007) Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer 109(9):1721–1728
Minami CA, Chung DU, Chang HR (2011) Management options in triple-negative breast cancer. Breast Cancer (Auckl) 5:175–199
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF (2007) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 25(1):118–145
Pao W, Girard N (2011) New driver mutations in non-small-cell lung cancer. Lancet Oncol 12(2):175–180
Sun Y, Ren Y, Fang Z, Li C, Fang R, Gao B, Han X, Tian W, Pao W, Chen H, Ji H (2010) Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol 28(30):4616–4620
Lee JW, Soung YH, Seo SH, Kim SY, Park CH, Wang YP, Park K, Nam SW, Park WS, Kim SH, Lee JY, Yoo NJ, Lee SH (2006) Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res 12(1):57–61
Teng YH, Tan WJ, Thike AA, Cheok PY, Tse GM, Wong NS, Yip GW, Bay BH, Tan PH (2011) Mutations in the epidermal growth factor receptor (EGFR) gene in triple negative breast cancer: possible implications for targeted therapy. Breast Cancer Res 13(2):R35
Bhargava R, Gerald WL, Li AR, Pan Q, Lal P, Ladanyi M, Chen B (2005) EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod Pathol 18(8):1027–1033
Burness ML, Grushko TA, Olopade OI (2010) Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J 16(1):23–32
Gumuskaya B, Alper M, Hucumenoglu S, Altundag K, Uner A, Guler G (2010) EGFR expression and gene copy number in triple-negative breast carcinoma. Cancer Genet Cytogenet 203(2):222–229
Kersting C, Tidow N, Schmidt H, Liedtke C, Neumann J, Boecker W, van Diest PJ, Brandt B, Buerger H (2004) Gene dosage PCR and fluorescence in situ hybridization reveal low frequency of egfr amplifications despite protein overexpression in invasive breast carcinoma. Lab Invest 84(5):582–587
Fukuyoshi Y, Inoue H, Kita Y, Utsunomiya T, Ishida T, Mori M (2008) EML4-ALK fusion transcript is not found in gastrointestinal and breast cancers. Br J Cancer 98(9):1536–1539
Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, Pandita A, Tang J, Modrusan Z (2009) Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res 7(9):1466–1476
Karnoub AE, Weinberg RA (2008) Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9(7):517–531
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417(6892):949–954
Gollamudi R, Ghalib MH, Desai KK, Chaudhary I, Wong B, Einstein M, Coffey M, Gill GM, Mettinger K, Mariadason JM, Mani S, Goel S (2010) Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs 28(5):641–649. doi:10.1007/s10637-009-9279-8
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP, Peters BA, Pujara K, Cordes S, Davis DP, Carlton VE, Yuan W, Li L, Wang W, Eigenbrot C, Kaminker JS, Eberhard DA, Waring P, Schuster SC, Modrusan Z, Zhang Z, Stokoe D, de Sauvage FJ, Faham M, Seshagiri S (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466(7308):869–873. doi:10.1038/nature09208
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP (1998) Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 4(7):844–847
Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, Nielsen TO (2008) Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res 14(5):1368–1376
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350(21):2129–2139
Schlomm T, Iwers L, Kirstein P, Jessen B, Kollermann J, Minner S, Passow-Drolet A, Mirlacher M, Milde-Langosch K, Graefen M, Haese A, Steuber T, Simon R, Huland H, Sauter G, Erbersdobler A (2008) Clinical significance of p53 alterations in surgically treated prostate cancers. Mod Pathol 21(11):1371–1378
Hirsch FR, Varella-Garcia M, Bunn PA Jr, Di Maria MV, Veve R, Bremmes RM, Baron AE, Zeng C, Franklin WA (2003) Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 21(20):3798–3807
Varella-Garcia M, Diebold J, Eberhard DA, Geenen K, Hirschmann A, Kockx M, Nagelmeier I, Ruschoff J, Schmitt M, Arbogast S, Cappuzzo F (2009) EGFR fluorescence in situ hybridisation assay: guidelines for application to non-small-cell lung cancer. J Clin Pathol 62(11):970–977
Boyault S, Drouet Y, Navarro C, Bachelot T, Lasset C, Treilleux I, Tabone E, Puisieux A, Wang Q (2012) Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res Treat 132:29–39
De Greve JL, Teugels E, De Mey J, Geers C, Galdermans D, Decoster L, In′t Veld P, Schallier D, Taton M, Shahidi M (2009) Clinical activity of BIBW 2992, an irreversible inhibitor of EGFR and HER2 in adenocarcinoma of the lung with mutations in the kinase domain of HER2neu. J Thorac Oncol 4:S307 (abstr.)
Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL (2006) HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10(1):25–38
Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA Jr, Varella-Garcia M (2005) Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 97(9):643–655
Hirsch FR, Herbst RS, Olsen C, Chansky K, Crowley J, Kelly K, Franklin WA, Bunn PA Jr, Varella-Garcia M, Gandara DR (2008) Increased EGFR gene copy number detected by fluorescent in situ hybridization predicts outcome in non-small-cell lung cancer patients treated with cetuximab and chemotherapy. J Clin Oncol 26(20):3351–3357
Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA (2005) Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med 353(2):133–144
Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S (2009) Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol 6(6):352–366
Lee JW, Soung YH, Kim SY, Park WS, Nam SW, Lee JY, Yoo NJ, Lee SH (2005) Absence of EGFR mutation in the kinase domain in common human cancers besides non-small cell lung cancer. Int J Cancer 113(3):510–511
Uramoto H, Shimokawa H, Nagata Y, Ono K, Hanagiri T (2010) EGFR-activating mutations are not present in breast tumors of Japanese patients. Anticancer Res 30(10):4219–4222
Sanchez-Munoz A, Gallego E, de Luque V, Perez-Rivas LG, Vicioso L, Ribelles N, Lozano J, Alba E (2010) Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC Cancer 10:136
Nofech-Mozes S, Trudeau M, Kahn HK, Dent R, Rawlinson E, Sun P, Narod SA, Hanna WM (2009) Patterns of recurrence in the basal and non-basal subtypes of triple-negative breast cancers. Breast Cancer Res Treat 118(1):131–137
WHO (2003) Tumors of the breast. In: Tavassoli FA, Devilee P (eds) Pathology and genetics of tumours of the breast and female genital organs. World Health Organisation classification of tumours. IARC Press, Lyon, pp 9–112
Elston CW, Ellis IO (1991) Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19(5):403–410
Sobin LH, Gospodarowicz MK, Wittekind C (2010) International Union against cancer (UICC) TNM classification of malignant tumours, 7th edn. Wiley-Liss, New York
Acknowledgments
The authors appreciate the excellent technical support of Gabriele Rieck, Silvia Schnöger, Sascha Eghtessadi, Jana Hagemann, and Sina Dietrich.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Grob, T.J., Heilenkötter, U., Geist, S. et al. Rare oncogenic mutations of predictive markers for targeted therapy in triple-negative breast cancer. Breast Cancer Res Treat 134, 561–567 (2012). https://doi.org/10.1007/s10549-012-2092-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2092-7