
Abstract
Most of breast cancers are resistant to mammalian target of rapamycin complex 1 (mTORC1) inhibitors rapamycin and rapalogs. Recent studies indicate mTORC2 is emerging as a promising cancer therapeutic target. In this study, we compared the inhibitory effects of targeting mTORC1 with mTORC2 on a variety of breast cancer cell lines and xenograft. We demonstrated that inhibition of mTORC1/2 by mTOR kinase inhibitors PP242 and OSI-027 effectively suppress phosphorylation of Akt (S473) and breast cancer cell proliferation. Targeting of mTORC2 either by kinase inhibitors or rictor knockdown, but not inhibition of mTORC1 either by rapamycin or raptor knockdown promotes serum starvation- or cisplatin-induced apoptosis. Furthermore, targeting of mTORC2 but not mTORC1 efficiently prevent breast cancer cell migration. Most importantly, in vivo administration of PP242 but not rapamycin as single agent effectively prevents breast tumor growth and induces apoptosis in xenograft. Our data suggest that agents that inhibit mTORC2 may have advantages over selective mTORC1 inhibitors in the treatment of breast cancers. Given that mTOR kinase inhibitors are in clinical trials, this study provides a strong rationale for testing the use of mTOR kinase inhibitors or combination of mTOR kinase inhibitors and cisplatin in the clinic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase at the nexus between oncogenic phosphoinositide 3-kinase (PI3K)/Akt signaling and critical downstream pathways that drive cancer cell growth, survival and resistance to therapeutic agents [1, 2]. mTOR kinase exists in two complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTOR interacts with raptor, Lst8, FKBP38, Deptor and PRAS40 to form mTORC1, the sensitive target of rapamycin that phosphorylates downstream targets of S6 kinase 1 (p70 S6K1) and eukaryotic initiation factor 4E binding protein-1 (4E-BP1) and control the cap-dependent protein translation, or with rictor, Lst8, Sin1, Deptor and Protor to form mTORC2, which is insensitive to acute rapamycin treatment and phosphorylates Akt at Ser473. Together, these complexes coordinate a variety of processes that include protein translation, autophagy, proliferation, survival and metabolism in response to nutrient, oxygen, energy, stress and growth factor signals [3]. Consistent with its role as a growth-promoting pathway, mTOR signaling is dysregulated in 50 % of all human malignancies and is a major cancer drug target [4].
As the first generation mTOR inhibitor, rapamycin, and rapalogs (everolimus, temsirolimus) can slow the proliferation of cancer cell lines and have achieved some success in caner treatment [5]. Unfortunately, their overall efficacy as cancer therapeutics has been limited to a few rare cancers, including mantle cell lymphoma, renal cell carcinoma, and endometrial cancer [6, 7]. It is increasingly recognized that the mechanism of action of rapamycin as a partial mTOR inhibitor is not sufficient for achieving abroad and robust anticancer effect, at least when these agents are employed in a monotherapy setting [5]. The major drawbacks of rapalogs are: (1) S6K is exquisitely inhibited, yet the control of 4E-BP and mRNA translation is far less sensitive [8]; (2) mTORC2 activity is not acutely blocked; (3) there is a feedback loop between mTORC1 and Akt. Treatment with rapalogs results in elevated Akt activity through S6K1 and insulin receptor substrate-1 (IRS-1), which serves as a mechanism to enhance cell survival when mTORC1 is inhibited [9].
Because the mTOR kinase domain is important for rapamycin-sensitive and -insensitive functions, mTOR ATP-competitive inhibitors have been developed recently that are able to completely suppress both mTORC1/C2 complex-mediated signaling, there by suppressing the feedback activation of Akt [10–14]. Importantly, they have shown marked improvement of anti-tumor activity in vivo and in vitro and the effectiveness of these drugs in cancer treatment is currently being tested in clinical trials [7, 15, 16].
In order to realize the full clinical potential of mTOR kinase inhibitors, a greater understanding of the molecular and cellular mechanisms of action of these compounds is needed. Furthermore, recent studies in cancer biology indicate that mTORC2 is emerging as a promising therapeutic target because its activity is essential for the transformation and vitality of a number of cancer cell types [17]. It is important to determine the efficacy of targeting of mTORC2 across a broad range of tumor types. In this study, we compared the inhibitory effects of targeting of mTORC1 with mTORC2 on a variety of breast cancer cell lines and xenograft and demonstrate that targeted inhibition of mTORC2, but not mTORC1, prevents breast cancer cell migration and promotes apoptosis in vitro and in vivo.
Materials and methods
Reagents and antibodies
PP242 and OSI-027 were purchased from Active Biochemicals Co. (Hongkong, China); Rapamycin was from Sigma Chemical Co. (St. Louis, MO). The following antibodies were used: 4E-BP1, phospho-4E-BP1 (T37/46), phospho-S6 (S235/236), raptor and cleaved poly (ADP-ribose) polymerase (PARP) from Cell Signaling Inc (Beverly, MA); S6, actin, P-Akt (S473), mTOR, rictor, Akt from Santa Cruz Biotech (Santa Cruz, CA).
Cell culture
Breast cancer cell lines, MCF-7, T47D, Bcap-37, BT-549, MDA-MB-231, ZR-7530, and ZR-751 were obtained from Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). MCF-7 and T47D were cultured in glucose-free DMEM supplemented with 10 % fetal bovine serum (FBS), Bcap-37, BT-549, ZR-7530, and ZR-751 were cultured in methionine-free RPMI 1640 supplemented with 10 % FBS, MDA-MB-231 cells were fed with L-15 with 10 % FBS.
Cell proliferation and clone formation
2,000 cells/well were plated into 96-well plates and treated with PP242 (20 nM–5 μM), OSI027 (100 nM–5 μM), and rapamycin (50 nM) for 72 h. Cell proliferation was evaluated by Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instruction. For clone formation, 200 of MCF-7 or MDA-MB-231 cells/well were seeded in 12-well plates in medium contain 0.1–1 μM of PP242 or OSI-027 and 10 % FBS. The medium was changed every 2 days. The number of clones was counted and the clone formation rate was calculated 10 days later.
RNA interference
Human mTOR, raptor, and rictor-specific siRNA were chemically synthesized by GenePharma Co., Ltd (Shanghai, China). Target sequences of these siRNA are: mTOR (5′-GAGCCUUGUUGAUCCUUAA-3′); Raptor (5′-CGAGAUUGGACGACCAAAU-3′); rictor (5′-GACUAUCCAUAAUCCUUA-3′);. Cells were transfected with the siRNA at 60 % confluence using lipofectamine 2000 (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions.
Western blotting
Immunoblotting was performed as previously described [18, 19]. In brief, cells were lysed in a buffer containing 1 % SDS. Equal amounts of whole protein extract were resolved on SDS–polyacrylamide gel, transferred to a nitrocellulose membrane (Amersham Biosciences, Italy), probed overnight at 4 °C with antibodies and then revealed using the ECL Western Blotting Analysis System.
Cell migration assay
MCF-7 or MDA-MB-231 cells were seeded in six-well plates and transfected with negative control (NC), mTOR, raptor or rictor siRNA, and grown until 100 % confluence. Cells were then pretreated with 2 μg/ml of mitomycin C for 24 h in a minimum medium (containing 0.5 % FBS). After making a straight scratch by using a pipette tip, cells were incubated in medium containing 0.5 % FBS in a 37 °C humidified incubator for 20 h and the wound distances were measured under a light microscope.
Apoptosis assays
Quantitation of apoptosis by propidium iodide staining was performed using an Apoptosis detection Kit (KeyGEN Biotech., Nanjin, China) according to the manufacturer’s instructions.
Drug combination analysis
Drug combination analysis was performed by using the method described by Chou and Talalay [20]. In brief, multiple drug dose–effect calculations and the combination index plots were generated using Calcusyn 2.1 software (Biosoft, Cambridge, UK). The general equation for the classic isobologram is given by CI = (D)1/(Dx)1 + (D)2/(Dx)2 + ((D)1 (D)2)/((Dx)1˙(Dx)2), where (Dx)1 and (Dx)2 in the denominators are the doses (or concentrations) for D1 (drug 1) and D2 (drug 2) alone that give x% apoptotic cells, whereas (D)1 and (D)2 in the numerators are the doses of drug 1 and drug 2 in combination that also induced x% cell apoptosis. CI <1 and CI < 0.3 indicated synergism and strong synergism, whereas CI = 1 and CI > 1 indicated additive effect and antagonism, respectively. CI/effect curves represent the CI versus the fraction (0–1) of cells killed by drug combination. Drug combination studies were performed using the following concentrations: 200 nM cisplatin and 20–400 nM of rapamycin, PP242 or OSI-027 were used in MCF-7, MDA-MB-231 or Bcap37 cell lines.
In vivo studies
Four-week-old female BALB/c nude mice were purchased from Guangdong Medical Experiment Animal Centre (Guangzhou, China). Each animal was injected with 1 × 107/0.1 ml of MDA-MB-231 cell suspension into the flanks. One week later, the mice were randomized into three groups (n = 5) and treated 7 days a week for vehicle control, rapamycin (1 mg/kg) or PP242 (10 mg/kg) solved in water containing 5 % 1-methyl-2-pyrrolidinone and 15 % polyvinylpyrrolidone. Bidimensional tumor measurements were taken every other day. At the end of the experiment on day 20 post-injection, mice were killed and tumors were removed, weighed, and then extracted for protein analysis, or were formalin fixed and paraffin embedded, and subjected to TUNEL staining using an apoptosis detection kit (KeyGEN Biotech. Nanjin, China). The percent of apoptotic cells was calculated under a microscope. All animal procedures were done in the nude mouse facility using protocols approved by the Animal Care and Use Committee of Southern Medical University.
Statistical analysis
The results were reported as mean ± SD. Statistical analyses were performed with one way ANOVA, and p < 0.05 was considered statistically significant.
Results
Targeted inhibition of mTORC1/2 signaling by mTOR kinase inhibitors PP242 and OSI-027 effectively suppresses breast cancer cell proliferation
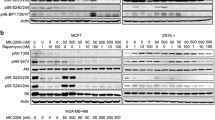
We first examined the distinct effects of targeting of mTORC1 by rapamycin and targeting of mTORC1/2 by two active-site mTOR inhibitors PP242 and OSI-027 on mTORC1/2 outputs in breast cancer MCF-7, T47D, MDA-MB-231, and Bcap-37 cell lines. PP242 and OSI-027 dose-dependently (50–500 nM) suppressed phosphorylation of Akt (S473), a mTORC2 phosphorylation site in all tested cell lines, whereas rapamycin treatment caused an increase in phosphorylation of Akt (S473) in MCF-7, MDA-MB-231, and Bcap-37 cells (Fig. 1a, b, c, d). Although all drugs effectively suppressed phosphorylation of S6 (S235/236), only mTOR kinase inhibitors PP242 effectively decreased the phosphorylation of 4E-BP1 (T37/46) in these cells (Fig. 1a, b, c, d). It is indicated that mTOR kinase inhibitors profoundly diminish mTORC1 and mTORC2 signaling, whereas rapamycin suppresses mTORC1 and enhances phosphorylation of Akt (S473) in some breast cancer cell lines.

Targeted inhibition of mTORC1/2 signaling by mTOR kinase inhibitors PP242 and OSI-027 in breast cancer cells. a MCF-7, b T47D, c MDA-MB-231 or d Bcap-37 cells were incubated with 50 nM rapamycin or indicated doses of PP242 or OSI-027 for 30 min. Cell lysates were then subjected to immunoblotting for levels of Phospho-Akt (S473), Akt, phospho-S6 (S235/236), S6, phospho-4E-BP1 (T37/46) and 4E-BP1
Second, the anti-proliferative activities of PP242 and OSI-027 compared with rapamycin in seven breast cancer cell lines were tested. We found that both targeting of mTORC1 (rapamycin) or dual targeting of mTORC1/2 (PP242 or OSI-027) effectively suppressed proliferation in the tested breast cancer cells (Supplementary Fig. 1a, b). Furthermore, PP242 and OSI-027 inhibited clone formation in a dose-dependent manner in MCF-7 and MDA-MB-231 cells (Supplementary Fig. 2).
Targeting of mTORC2 but not mTORC1 promotes apoptosis in breast cancer cells
Akt represents an important intracellular survival signaling under a variety of conditions [21]. Rapamycin induced feedback activation of Akt in MCF-7, MDA-MB-231, and Bcap-37 breast cancer cells (Fig. 1). Accordingly, rapamycin did not exhibit any pro-apoptotic activity in these cells (Fig. 2a, b, c, d). The Akt activation induced by loss of feedback inhibition could be prevented by mTOR kinase inhibitors PP242 and OSI-027 (Fig. 1). In consistent with the inhibitory activities of PP242 and OSI-027 in Akt (S473) phosphorylation (Fig. 1), these drugs significantly enhanced cleavage of poly (ADP-ribose) polymerase (PARP) and number of apoptotic cells in serum-starved MCF-7, MDA-MB-231, and Bcap-37 cell lines (Fig. 2a, b, c, d).

PP242 and OSI-027, but not rapamycin promote apoptosis in breast cancer cells. a MCF-7, b MDA-MB-231 or c Bcap-37 cells were incubated with 100 nM rapamycin, or 200 nM of PP242 or OSI-027 for 36 h in serum-free medium. Apoptotic cell death was then quantified using PI staining. **p < 0.01 compared with control and rapamycin treatment. d MCF-7, MDA-MB-231 or Bcap-37 cells were incubated with 100 nM rapamycin, or 200 nM of PP242 or OSI-027 for 24 h, cell lysates were subjected to immunoblotting for levels of cleaved PARP and actin
To further identify the roles of mTORC1 and mTORC2 in breast cancer cell survival, the effects of raptor, mTOR or rictor knockdown on apoptosis were examined. Raptor, rictor or mTOR siRNA markedly decreased protein levels of raptor, rictor or mTOR and reduced the phosphorylation of their outputs S6 (S235/236) and Akt (S473), respectively in MCF-7 and MDA-MB-231 cells (Fig. 3a). Knockdown of rictor and mTOR, but not raptor promoted cleavage of PARP and increased the apoptotic cells in serum-starved MDA-MB-231 cells (Fig. 3a, b). Taken together, it is suggested that targeting of mTORC2 but not mTORC1 promotes apoptosis in breast cancer cells.

Rictor or mTOR knockdown, but not raptor knockdown potentiates apoptosis in breast cancer cells. a MDA-MB-231 cells were transfected with negative control (NC), mTOR, raptor or rictor siRNA for 48 h, subsequently serum-starved for another 36 h. cell lysates were subjected to immunoblotting for levels of mTOR, raptor, rictor, phospho-Akt (S473), Akt, phospho-S6 (S235/236), S6, cleaved PARP and actin. b Apoptotic cell death was quantified using apoptosis detection Kit. *p < 0.01 compared with NC and raptor siRNA transfected cells
Inhibition of mTORC2 potentiates cisplatin-induced apoptosis in breast cancer cells
Cisplatin, a chemotherapeutic agent that is commonly used for many cancers, could induce apoptosis in a variety of cancer cell lines [22]. Drug combination index (CI) analysis is a generalized method for analyzing the effects of multiple drugs and for determining summation, synergism, and antagonism. The CI method helps answer whether there are synergism with two drugs, how much the two drugs synergism and symergism at what dose levels. The synergy of two drugs depend on the potency and the shape of the dose–effect curve of each drug, then we can determine the effectives of synergism, antagonism and quantitatively, by using the CI equation. To examine the interactions of mTOR kinase inhibitors with low dose of cisplatin, dose–response studies were conducted and CI values were calculated with the CalcuSyn program in MCF-7, MDA-MB-231, and Bcap-37 cells. As shown in Fig. 4, when PP242 or OSI-027 was applied at low concentrations (20–400 nM), the CI values were <1.0, indicating synergistic interactions between PP242 or OSI-027 and low dose cisplatin (200 nM) (Fig. 4a, b). On the contrary, rapamycin did not show any synergistic interactions with cisplatin, as the CI values were >1.0 (Fig. 4c). Furthermore, PP242 or OSI-027, but not rapamycin notably enhanced cisplatin-induced cleavage of PARP (Fig. 4d). Most importantly, rictor or mTOR knockdown, but not raptor knockdown, markedly enhanced cisplatin-induced cleavage of PARP and the number of apoptotic cells in MDA-MB-231 cells (Fig. 5a, b and Supplementary Fig. 3). Taken together, our results indicate that targeted inhibition of mTORC2 but not mTORC1 markedly potentiates cisplatin-induced apoptosis in breast cancer cells.

PP242 and OSI-027, but not rapamycin has synergistic interactions with low dose of cisplatin. a MCF-7, b MDA-MB-231 or c Bcap37 cells were treated with 200 nM cisplatin and 20–400 nM of rapamycin, PP242 or OSI-027 for 36 h. Apoptotic cell death was then quantified using PI staining and CI values were calculated with the CalcuSyn program. d MDA-MB-231 cells were treated with 200 nM of rapamycin, PP242 or OSI-027 combined with or without 200 nM cisplatin for 36 h, cells were lysed and subjected to immunoblotting for levels of cleaved PARP and actin

Rictor and mTOR knockdown, but not raptor knockdown potentiates cisplatin-induced breast cancer cell apoptosis. a MDA-MB-231 cells were transfected with negative control (NC), mTOR, raptor or rictor siRNA for 48 h, subsequently treated with 200 nM of cisplatin for another 36 h. Apoptotic cell death was then quantified using PI staining. *p < 0.01 compared with no cisplatin treatment; # p < 0.01 compared with NC and raptor siRNA transfected cells. b Cells were lysed and subjected to immunoblotting for levels of mTOR, raptor, rictor, phospho-Akt (S473), Akt, phospho-S6 (S235/236), S6, cleaved PARP and actin
Targeted inhibition of mTORC2 prevents breast cancer cell migration
Metastasis is the major cause of mortality and morbidity among breast cancer patients. Invasion of cancer cells into surrounding tissue and the vasculature is an initial step in tumor metastasis. This requires migration of cancer cells [23]. To investigate different roles of mTORC1 and mTORC2 in breast cancer cell migration, effects of targeted inhibition of mTORC1 or mTORC2 on cell migration were examined by using a wound healing assay in a serum-free medium. As shown in Fig. 6a and b and Supplementary Fig. 4, PP242- or OSI-027- treated and mTOR or rictor siRNA transfected MCF-7 cells filled the gap more slowly than vehicle control or rapamycin- treated or raptor siRNA transfected MCF-7 cells did, suggesting that inhibition of mTORC2 but not mTORC1 prevented cell migration. These results were repeated in MDA-MB-231 cells (Fig. 6c and d, Supplementary Fig. 5), further confirmed the critical role of mTORC2 in breast cancer cell migration.

Targeted inhibition of mTORC2 prevents breast cancer cell migration. a MCF-7 were pretreated with 2 μg/ml mitomycin C for 24 h followed by incubation with 100 nM of rapamycin, PP242 or OSI-027, or b transfected with negative control (NC), mTOR, raptor or rictor siRNA for 48 h, subsequently pretreated with 2 μg/ml mitomycin C for 24 h and subjected to wound healing assay as described in “Materials and methods” section. The wound distances were measured under a light microscope 20 h after wounding. c MDA-MB-231 cells were treated as in a, or d transfected with negative control, mTOR, raptor or rictor siRNA for 48 h. Cells were then treated with 2 μg/ml mitomycin C for 24 h and subjected to wound healing assay. *p < 0.05 compared with vehicle and rapamycin-treated cells, or compared with NC and raptor siRNA transfected cells
In vivo administration of PP242 but not rapamycin as a single agent effectively prevents breast tumor growth and induces apoptosis in xenograft
To compare the in vivo efficacy of inhibition of mTORC1 with dual inhibition of mTORC1/2, nude mice bearing MDA-MB-231 breast tumor xenografts were dosed with rapamycin or PP242. The PP242 therapy as single agent markedly inhibited breast tumor growth (tumor areas and tumor weight) while rapamycin was ineffective to suppress the tumor growth (Fig. 7a, b). Most importantly, in vivo administration of PP242 induced significant apoptosis in the breast tumor xenograft (Fig. 8a, b). But rapamycin treatment failed to induce significant apoptosis in this model. Western blotting analysis demonstrated that PP242 inhibited the putative targets of both mTORC1 and mTORC2 signaling in vivo, as evidenced by the inhibition of phosphorylated Akt (S473) and S6 (S235/236) (Fig. 8c). In consistent with in vitro results, rapamycin suppressed mTORC1 (P-S6) but not mTORC2 (P-Akt) (Fig. 8c). Our results indicate that dual targeting of mTORC1/2 but not mTORC1 effectively suppresses breast tumor growth and induces apoptosis in vivo.

In vivo administration of PP242 but not rapamycin as single agent effectively prevents breast tumor growth in xenografts. a Tumor growth curves of MDA-MB-231 xenografts in nude beige mice treated with saline, rapamycin, or PP242 daily by gavage. Each treatment group comprised five mice. Each data point means estimated areas of the tumors. b Tumor weight of MDA-MB-231 xenografts in nude mice treated with saline, rapamycin, or PP242. *P < 0.05 for comparison of PP242 therapy versus control or rapamycin therapy

In vivo administration of PP242 but not rapamycin as single agent induces cell apoptosis in xenografts. a and b MDA-MB-231 xenografts in nude beige mice treated with saline, rapamycin, or PP242 were stained with TUNEL apoptosis detection kit and the percent of apoptotic cells was calculated under a microscope. *P < 0.05 for comparison of PP242 therapy versus control or rapamycin therapy. c Activities of mTORC1 and mTORC2 in MDA-MB-231 xenografts from mice treated with saline, rapamycin, or PP242
Discussion
In this study, we showed that targeted inhibition of mTORC2, but not mTORC1, prevents migration and promote apoptosis in breast cancer cell lines in vitro and breast tumor xenograft in vivo. We also have demonstrated the synergistic antitumor activity achieved by combining mTOR kinase inhibitors with cisplatin. Our data suggest that agents that inhibition of either mTORC2 or mTORC1/2 may have advantages over selective mTORC1 inhibitors in the treatment of breast cancers. Given that mTOR kinase inhibitors (e.g., OSI-027) are in clinical trials, this study provides a strong rationale for testing the use of mTOR kinase inhibitors or combination of mTOR kinase inhibitors with cisplatin in the clinic.
The recent development of mTOR kinase inhibitors not only provide invaluable tools for investigating the complex mTOR signaling network but also offer considerable new opportunities to exploit fully the therapeutic potential of mTOR targeting in cancer [10–12]. Some mTORC1/2 kinase inhibitors such as Torin1, PP242, PP30, Ku-0063794, OSI-027, ADZ2014, INK128, WAY-600, WAY-687, WAY-354, and AZD8055 have been tested in some in vitro and in vivo pre-clinical models and shed great promise for anticancer therapy and some of them are rapidly moving into clinical trials [7, 15, 16, 24]. However, we need a greater understanding of the mechanism of action of mTORC1/2 inhibitors in cancer cells, such as which cellular processes regulated by mTORC1 and/or mTORC2 are relevant to the therapeutic effects. In the current study, we demonstrated that although either rapamycin, or PP242 and OSI-027 could suppress proliferation in a variety of breast cancer cell lines, PP242 but not rapamycin effectively prevented breast tumor growth in vivo. Moreover, inhibition of mTORC2 or mTORC1/2, but not mTORC1 potentiated apoptosis and suppressed cell migration. These data suggest that promotion of apoptosis and suppression of cell migration may contribute to the therapeutic effects of mTORC1/2 inhibitors. It will also be important to identify biomarkers of drug efficacy and resistance, subsequently predict what cancer patients will benefit from these inhibitors. Two recent studies have demonstrated that breast cancer cells with PIK3CA and/or HER2 mutations, but not PTEN loss of function, are sensitive to PP242 or PI3K/mTOR dual inhibitory drug NVPBEZ235 [25, 26]. We also found that the proliferation of cells such as MCF-7 and T47D (with PI3KCA mutations) is sensitive, while MDA-MB-231(with wild type PIK3CA and wild type PTEN) is less sensitive to PP242 and OSI-027. However, MDA-MB-231 is sensitive to rictor knockdown- and mTORC1/2 inhibitors-potentiated cell apoptosis and suppressed cell migration, suggesting a critical role of mTORC2 in later phases of breast cancer progression. Another need is to test mTORC1/2 kinase inhibitors in combination with current therapies. In a renal cell carcinoma model the compound WYE-132 showed greater ability than CCI-779 to synergize with bevacizumab (Avastin), a monoclonal antibody to vascular endothelial growth factor (VEGF-A) [11]. Although cisplatin is not commonly used for breast cancer, it becomes a potential tool in the management of some types of breast tumor such as basal-like breast cancer [27]. Our finding that mTORC1/2 inhibitors markedly potentiate cisplatin-induced apoptosis in breast cancer cells suggests the clinical potential of mTORC1/2 kinase inhibitors for combinatorial therapies. Careful strategies will need to be employed when developing a dosing regimen to best obtain the full therapeutic benefit of drug combinations in breast cancer [28–30].
Recent studies suggest that mTORC2 activity is essential for the transformation and vitality of a number of cancers driven by mutations of PI3K or loss of PTEN. Glioma cell lines and tissues exhibit rictor overexpression, which results in elevated mTORC2 activity and promotes anchorage-independent growth, cellular motility, and in vivo growth [31]. Prostate cancers lacking PTEN require mTORC2 to form tumors when injected into nude mice. The development of prostate cancer caused by PTEN deletion in prostate epithelium required mTORC2, whereas mTORC2 activity is not essential for maintaining the integrity of normal prostate epithelium [32]. Although both mTORC1 and mTORC2 have been reported to mediate epithelial–mesenchymal transition (EMT) and cell motility in epithelial, colon cancer, and podocytes [33–35], we found that inhibition of mTORC2 but not mTORC1 promotes apoptosis and suppresses migration in breast cancer cells, indicating the critical role of mTORC2 in breast cancer cell survival and migration. Another report also suggested that rictor may interact with PKCζ and regulate cancer metastasis [36]. Although existing ATP-competitive mTOR inhibitors may prevent mTORC2 activity, the concurrent inhibition of mTORC1 might introduce hyper-activation of PI3K signaling and possible deleterious effects to normal host tissue, which will limit their therapeutic potential [37]. Thus, mTORC2-specific inhibitor may be a promising therapeutic agent for certain cancers [17]. The relative importance of mTORC1 versus mTORC2 inhibition for suppression of cancer cell proliferation and survival is not yet clear, and might be dependent on cell context. If mTORC2 inhibition contributes to the mechanism, it will be important to determine which mTORC2 substrates are the relevant mediators of cancer cell growth and survival.
References
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12(1):21–35
Guertin DA, Sabatini DM (2005) An expanding role for mTOR in cancer. Trends Mol Med 11(8):353–361
Laplante M, Sabatini DM (2009) mTOR signaling at a glance. J Cell Sci 122(Pt 20):3589–3594
Janes MR, Fruman DA (2010) Targeting TOR dependence in cancer. Oncotarget 1(1):69–76
Benjamin D, Colombi M, Moroni C, Hall MN (2011) Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 10(11):868–880
Houghton PJ (2010) Everolimus. Clin Cancer Res 16(5):1368–1372
Guertin DA, Sabatini DM (2009) The pharmacology of mTOR inhibition. Sci Signal 2(67):pe24
Thoreen CC, Sabatini DM (2009) Rapamycin inhibits mTORC1, but not completely. Autophagy 5(5):725–726
Carew JS, Kelly KR, Nawrocki ST (2011) Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol 6(1):17–27
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM (2009) Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 7(2):e38
Yu K, Shi C, Toral-Barza L, Lucas J, Shor B, Kim JE, Zhang WG, Mahoney R, Gaydos C, Tardio L, Kim SK, Conant R, Curran K, Kaplan J, Verheijen J, Ayral-Kaloustian S, Mansour TS, Abraham RT, Zask A, Gibbons JJ (2010) Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res 70(2):621–631
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284(12):8023–8032
Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA (2010) Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 16(2):205–213
Shao H, Gao C, Tang H, Zhang H, Roberts LR, Hylander BL, Repasky EA, Ma WW, Qiu J, Adjei AA, Dy GK, Yu C (2012) Dual targeting of mTORC1/C2 complexes enhances histone deacetylase inhibitor-mediated anti-tumor efficacy in primary HCC cancer in vitro and in vivo. J Hepatol 56(1):176–183
Zhang YJ, Duan Y, Zheng XF (2011) Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today 16(7–8):325–331
Schenone S, Brullo C, Musumeci F, Radi M, Botta M (2011) ATP-competitive inhibitors of mTOR: an update. Curr Med Chem 18(20):2995–3014
Sparks CA, Guertin DA (2010) Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 29(26):3733–3744
Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y (2007) Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318(5852):977–980
Li M, Zhao L, Liu J, Liu A, Jia C, Ma D, Jiang Y, Bai X (2010) Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell Signal 22(10):1469–1476
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9(1):59–71
Cohen SM, Lippard SJ (2001) Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol 67:93–130
Li DM, Feng YM (2011) Signaling mechanism of cell adhesion molecules in breast cancer metastasis: potential therapeutic targets. Breast Cancer Res Treat 128(1):7–21
Babcock JT, Quilliam LA (2011) Rheb/mTOR activation and regulation in cancer: novel treatment strategies beyond rapamycin. Curr Drug Targets 12(8):1223–1231
Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, Wang S, Garcia-Echeverria C, Maira SM (2009) Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci USA 106(52):22299–22304
Weigelt B, Warne PH, Downward J (2011) PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 30(29):3222–3233
Wong SW, Tiong KH, Kong WY, Yue YC, Chua CH, Lim JY, Lee CY, Quah SI, Fow C, Chung C, So I, Tan BS, Choo HL, Rosli R, Cheong SK, Leong CO (2011) Rapamycin synergizes cisplatin sensitivity in basal-like breast cancer cells through up-regulation of p73. Breast Cancer Res Treat 128(2):301–313
O’Regan R, Hawk NN (2011) mTOR inhibition in breast cancer: unraveling the complex mechanisms of mTOR signal transduction and its clinical implications in therapy. Expert Opin Ther Targets 15(7):859–872
Awada A, Cardoso F, Fontaine C, Dirix L, De Greve J, Sotiriou C, Steinseifer J, Wouters C, Tanaka C, Zoellner U, Tang P, Piccart M (2008) The oral mTOR inhibitor RAD001 (everolimus) in combination with letrozole in patients with advanced breast cancer: results of a phase I study with pharmacokinetics. Eur J Cancer 44(1):84–91
deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M (2004) Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res 10(23):8059–8067
Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J (2007) mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res 67(24):11712–11720
Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM (2009) mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15(2):148–159
Shorning BY, Griffiths D, Clarke AR (2011) Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial–mesenchymal transition in the mouse bladder. PLoS ONE 6(1):e16209
Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, Lee EY, Weiss HL, O’Connor KL, Gao T, Evers BM (2011) mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res 71(9):3246–3256
Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Ruegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL (2011) mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121(6):2181–2196
Zhang F, Zhang X, Li M, Chen P, Zhang B, Guo H, Cao W, Wei X, Cao X, Hao X, Zhang N (2010) mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res 70(22):9360–9370
Proud CG (2011) mTOR signalling in health and disease. Biochem Soc Trans 39(2):431–436
Acknowledgments
The study was supported by the National Natural Sciences Foundation of China (30900555 and 91029727), Program for Changjiang Scholars and Innovative Research Team in University (IRT1142) and The State Key Development Program for Basic Research of China (2009CB 918904).
Conflict of interest
The authors declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Haiyan Li, Jun Lin and Xiaokai Wang contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Li, H., Lin, J., Wang, X. et al. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res Treat 134, 1057–1066 (2012). https://doi.org/10.1007/s10549-012-2036-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2036-2