
Abstract
We investigated the role of human natural killer (NK) cells in the peripheral blood (PB) and liver in controlling breast cancer. The proportion of NK cells among liver mononuclear cells was significantly higher than among PB mononuclear cells. Liver NK cells inductively expressed higher levels of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) than PB NK cells in response to interleukin-2 (IL-2). Liver NK cells displayed higher cytotoxicity against various breast cancer cell lines (MDA-MB231, MDA-MB453, MDA-MB468, and MCF-7) after IL-2 stimulation than did PB NK cells. Anti-HER2 monoclonal antibody (mAb) promoted the cytotoxicity of both the types of NK cells toward HER2-expressing cell lines. All breast cancer cell lines highly expressed death-inducing TRAIL receptors, death receptor 4, but did not express death-inhibitory receptors (DcR1 and DcR2). Both PB and liver NK cell-induced cytotoxicity was inhibited partially by anti-TRAIL mAb and more profoundly by the combination of anti-TRAIL mAb and concanamycin A, indicating that TRAIL and perforin are involved. IL-2-stimulated liver and PB NK cells exhibited upregulated expression of CXCR3, which bind to the chemokines CXCL9, CXCL10, and CXCL11 secreted by breast cancer cells. We also found that IFN-γ promoted the production of CXCL10 from breast cancer cells. The results of this study show that IFN-γ secreted from NK cells likely promotes the production of CXCL10 from breast cancer cells, which in turn accelerates the migration of CXCR3-expressing NK cells into the tumor site. These findings suggest the possibility of a therapeutic approach by either activation of endogenous PB and liver NK cells or adoptive transfer of in vitro-activated autologous NK cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Natural killer (NK) cells, the frontline defense in cellular immunity, exert an effector function on neoplastic cells, modified cells, and invading infectious microbes without the necessity for priming [1, 2]. Although, NK cells might play an important role in prevention of both early and metastatic cancer, the role of NK cell activity in controlling breast cancer is still controversial and few studies have addressed whether enhancing this activity is of clinical benefit to breast cancer patients.
A variety of mechanisms are involved in controlling neoplastic cells by NK cells, one of which is the direct release of cytolytic granules that contain perforin, granzymes, and granulysin by exocytosis to kill target cells (i.e., the granule exocytosis pathway) [3, 4]. Most mature human NK cells in peripheral blood (PB) constitutively express granzyme B and perforin, and have basal cytotoxicity against NK-sensitive targets. Cytokine exposure with interleukin (IL)-2 or IL-15 is known to increase the baseline granzyme B and perforin abundance and cytotoxic activity of NK cells, and also converts basal NK cytotoxicity to lymphokine-activated killing. Another mechanism is mediated by death-inducing ligands such as Fas ligand (FasL) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL). Fas, a TNF family protein, is expressed on breast cancer cell membranes [5, 6], suggesting that activation of the Fas/FasL pathway induces apoptosis mediated by caspase activation. An additional mechanism is involved when HER2-overexpressing breast cancer cells are targeted because differential levels of HER2 expression in normal versus HER2-overexpressing tumor cells, together with the clear involvement of HER2 in tumor progression, make HER2 an ideal target for therapeutic approaches. NK cell-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) is thought to contribute to the therapeutic effects of monoclonal antibodies (mAbs) specifically directed against the extracellular domain of HER2 (trastuzumab).
NK cells are abundant in the liver in contrast to their relatively small percentage in the peripheral lymphatics and other lymphatic organs in rodents [7–9] and humans [10]; however, the underlying reason for the anatomically biased distribution of NK cells has not been elucidated. In addition, liver NK cells have been shown to mediate higher cytotoxic activity against tumor cells than PB NK cells do in rodents [7–9, 11]. However, these functional differences between PB and liver NK cells have not been extensively investigated in humans because of the limited availability of appropriate samples. In this study, we extracted NK cells from allograft liver perfusates in clinical liver transplantation and examined the quantitative and qualitative cytotoxic functions of those liver NK cells targeting various breast cancer cell lines in comparison with PB NK cells. Through the experiments, we attempted to define whether PB NK cells can recognize and kill breast cancer cells, and whether liver resident NK cells can hinder metastasis of breast cancer to the liver, to assess the potential therapeutic use of NK cells, i.e., by either activation of endogenous NK cells or adoptive transfer of in vitro-activated autologous NK cells. As the therapeutic efficacy of endogenous or exogenous NK cells likely depends on their migration and accumulation at tumor metastasis sites, we further analyzed the expression of receptors and ligands for chemokines secreted from breast cancer cells on PB and liver NK cells.
Materials and methods
Isolation of liver and PB lymphocytes
Liver mononuclear cells (LMNCs) were obtained from liver perfusates in clinical living donor liver transplantation as previously described [10]. LMNCs were isolated by gradient centrifugation with Separate-L (Muto Pure Chemicals Co., Ltd, Tokyo, Japan). PB mononuclear cells (PBMCs) were also isolated by gradient centrifugation with Separate-L from heparinized PB from healthy volunteers and liver transplant donors. LMNCs and PBMCs were suspended in DMEM (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated fetal calf serum (FCS) (Sanko Chemical Co., Tokyo, Japan), 25 mmol/l HEPES buffer (Gibco), 50 μmol/l 2-mercaptoethanol (Katayama Chemical Co., Osaka, Japan), 50 U/ml penicillin, and 50 μg/ml streptomycin (Gibco) (10% DMEM). The ethics committee at Hiroshima University Hospital approved this study.
Cell culture
LMNCs and PBMCs were cultured with human recombinant IL-2 (100 Japanese reference U/ml; Takeda, Tokyo, Japan) in DMEM at 37°C in a 5% CO2 incubator. Cells were harvested for further analyses after 5 days in culture. Cell viability was assessed by the dye-exclusion test.
Isolation of NK cells
LMNCs and PBMCs were separated into a CD3−CD56+ NK cell fraction and a non-NK cell fraction (T cells, NKT cells, B cells, and monocytes/macrophages) by magnetic cell sorting (Miltenyi Biotec, Bergisch Gladbach, Germany), using the human NK cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. The purity of isolated fractions was assessed by FCM, and only preparations with purities >90% were used for functional studies.
Cell lines
The human breast cancer cell lines were obtained as follows: MDA-MB-231 and MDA-MB-468 were from ATCC (Manassas, VA), MDA-MB-453 was from Riken Cell Bank (Saitama, Japan), and MCF-7 was kindly provided by Dr. A. Kikuchi (Osaka University, Suita, Japan). MDA-MB-231, MDA-MB-453, and MDA-MB468 cells were cultured in Leibovitz’s L-15 medium (Invitrogen) supplemented with 10% FCS at 37°C in a 0% CO2 incubator. MCF-7 cells were cultured in DMEM supplemented with 10% FCS at 37°C in a 5% CO2 incubator. The phenotypic features of those cell lines were well documented [12, 13].
Flow cytometric analyses
Flow cytometric analyses were performed using a FACSCalibur® cytometer (BD Biosciences, San Jose, CA). The following mAbs were used for surface staining of the lymphocytes: anti-CD3-fluorescein isothiocyanate (FITC), HIT3a; anti-CD56-phycoerythrin (PE), B159; anti-NKG2D-allophycocyanin (APC), 1D11; anti-CD183-APC, CXCR3 (1C6/CXR3); and anti-FasL-biotin, NOK-1. The biotinylated mAb was visualized using APC-streptavidin (all mAbs from BD Biosciences, Franklin Lakes, NJ).
To analyze activating receptors or TRAIL expression on NK cells, LMNCs and PBMCs were stained with anti-CD3-FITC, anti-CD56-APC, anti-TRAIL-PE, RIK-2; anti-NKp30-PE, P30-15; anti-NKp44-PE, P44-8.1; and anti-NKp46-PE, 9E2/NKp46; mAbs (all mAbs from BD Biosciences). To analyze inhibitory receptors on NK cells, LMNCs and PBMCs were stained with anti-CD3-APC, SP34-2; anti-CD56-PE, anti-CD-158a-FITC, HP-3E4; anti-CD-158b-FITC, CH-L; or anti-CD94-FITC, HP-3D9; mAbs (all mAbs from BD Biosciences). To analyze the expression of CD94/NKG2A or CD94/NKG2C on NK cells, LMNCs and PBMCs were stained with anti-CD3-PerCP, SK7; (BD Biosciences), anti-CD56-APC, anti-CD94-FITC, and anti-NKG2A-PE, z199; (Beckman Coulter), or anti-NKG2C-PE, 134591; (R&D Systems, Minneapolis, MN) mAbs. To analyze TRAIL receptors on breast cancer cell lines, they were stained with biotin-conjugated anti-TRAIL-R1/DR4, DJR1; anti-TRAIL-R2/DR5, DJR2-4; anti-TRAIL-R3/decoy receptor (DcR) 1, DJR3; or anti-TRAIL-R4/DcR2, DJR4-1; mAbs (all mAbs from eBioscience). To analyze HER2 expression on breast cancer cell lines, breast cancer cells were stained with PE-conjugated anti-HER2 mAb, Neu 24.7; (Becton–Dickinson). All biotinylated mAbs were visualized with APC-streptavidin (BD Biosciences). Dead cells were excluded from analysis by light-scatter and propidium iodide staining.
Immunohistochemistry
Surgically resected breast specimens were obtained from breast cancer patients who had undergone curative tumor resection at Hiroshima University Hospital. Breast specimens that had been pathologically proven to be normal (i.e., excluding mastopathic and mastitic tissues) were used as normal controls. Each tumor section (4-μm thickness) was deparaffinized and subjected to antigen retrieval by incubation in 10 mM citrate buffer (sodium citrate, pH 6.0) at 99°C for 25 min. To block non-specific antibody binding, sections were incubated with blocking solution (PBS containing Sanglopor I.V., 1 mg/ml; CSL Behring AG, Bern, Switzerland) for 20 min. Sections were then incubated overnight at 4°C in blocking solution in the presence of the first antibody (biotin-conjugated anti-TRAIL-R1/DR4, anti-TRAIL-R2/DR5, anti-TRAIL-R3/DcR1, and anti-TRAIL-R4/DcR2) (5 μg/ml) (all mAbs from eBioscience) or biotin-conjugated mouse IgG1, κ isotype control (P3) at the same concentration (eBioscience). Sections were washed twice in PBS, and primary antibody binding sites were visualized using the Dako EnVision Kit (Dako, Copenhagen, Denmark) according to the manufacturer’s instructions. Sections were faintly counterstained with Harris’ hematoxylin and mounted with glycerol gelatin.
Cytotoxicity assay
The 51Cr-labeled breast cancer cells were incubated in a total volume of 200 μl with effector cells in L-15 medium in round-bottomed 96-well microtiter plates (BD Falcon) for 4 h. IL-2-stimulated NK cells were used as effectors at an effector-to-target (E:T) ratio of 1.25:1–10:1. To evaluate the ADCC of trastuzumab, the assay was performed in the presence of 0.1 μg/ml trastuzumab (kindly provided by Genentech Inc., San Francisco, CA). Target cells were incubated either in culture medium alone to determine spontaneous 51Cr release or in a mixture of the culture medium and 2% Nonidet P-40 (Nakalai Tesque Inc., Kyoto, Japan) to determine the maximum 51Cr release for controls. The radioactivity of the cell-free supernatants was measured in a gamma counter. The percentage of specific 51Cr release was calculated as % cytotoxicity = ([cpm of experimental release − cpm of spontaneous release]/[cpm of maximum release − cpm of spontaneous release]) × 100. In some experiments, the 51Cr-labeled breast cancer cells were incubated with effector cells either for 4 or 18 h in the presence of 10 μg/ml of anti-TRAIL (N2B2) mAb, 10 μg/ml of anti-FasL (MFL3) mAb (both from BD Pharmingen), and/or 50 nmol/l of concanamycin A (CMA) (Wako Chemicals, Osaka, Japan), which inhibits perforin-mediated cytotoxicity [14].
Cytometric bead assay
Chemokine production in the cell culture supernatants was analyzed by the cytometric bead array (CBA) using Human MIG (CXCL9), Human IP-10 (CXCL10), and Human I-TAC (CXCL11) Flex Sets (BD Bioscience), according to the manufacturer’s instructions, for the production of CXCL9, CXCL10, and CXCL11, respectively.
Cell migration assay
Migration assays were performed in transwell culture inserts (BD Falcon) of 6.4-mm diameter and 3-μm pore filters. MDA-MB231 and MDA-MB468 cells (1 × 106/well) were cultured in the lower chamber of a 24-well plate (BD Falcon) in 0.5 ml L-15 medium. After 2 days, IL-2-activated NK cells derived from PBMCs and LMNCs in 0.2 ml L-15 medium were added to the upper chamber (1 × 106/well), and cells were allowed to migrate for 2 h.
Enzyme-linked immunosorbent assay
The supernatants from the cell migration assay and coculture of MDA-MB231 or MDA-MB468 with IFN-γ for 2 or 6 h were used. IFN-γ and CXCL10 levels in the cell culture supernatants were determined by an enzyme-linked immunosorbent assay (ELISA) with the Quantikine kit (R&D Systems), according to the manufacturer instructions. Absorbance was measured at 492 nm on a microplate reader (MTP-300; CORONA Electric, Ibaraki, Japan).
Statistical analysis
Data are presented as mean ± SEM. The statistical differences of the results were analyzed by the 2-tailed, paired t test and Mann–Whitney U test, using Excel. P values of <0.05 were considered statistically significant.
Results
Phenotypic properties of human NK cells
NK cells are abundant in the liver in contrast to their relatively small percentage in the peripheral lymphatics and other lymphatic organs in humans [10]. While NK cells in circulating lymphocytes have been phenotypically and functionally defined, those that reside in the liver remain to be characterized. We phenotypically analyzed the LMNCs that were extracted from the perfusates of allograft livers during liver transplantation surgery. The proportions of CD56+CD3− NK and CD56+CD3+ NKT cells in the LMNCs extracted from liver perfusates were significantly higher than those in the PBMCs (Fig. 1a). Although this non-destructive method might allow some extent of contamination with circulating mononuclear cells, these data were consistent with previous reports using the enzymatic dissociation method [15]. Among CD56+CD3− NK cells, CD56bright cells, which constitutively expressed TRAIL, were abundant in LMNCs but were almost undetectable in PBMCs (Fig. 1b–d). On effector molecule analyses, the expression of TRAIL was significantly upregulated in both liver and PB NK cells after cultivation with IL-2 for 5 days. Both the proportion of the TRAIL+ fraction and the staining intensity of liver NK cells were significantly higher than those of PB NK cells (Fig. 2a). Neither PB nor liver NK cells expressed FasL even after IL-2 stimulation (Fig. 2b). We further analyzed the C-type lectin-like receptors CD94, NKG2A, and NKG2C and killer cell immunoglobulin-like receptors (KIR) such as CD158a and CD158b (Fig. 3). CD94 recognizes the non-classical MHC class Ib molecule HLA-E, whereas KIRs are MHC class I-restricted molecules that recognize HLA-A, HLA-B, HLA-C, and HLA-G molecules. All freshly isolated PB and liver NK cells expressed CD94, and cell subpopulations expressed CD158a/CD158b (Fig. 3). No statistically significant differences were observed in the expression of CD94, NKG2A, NKG2C, CD158a, and CD158b between PB and liver NK cells. IL-2 stimulation maintained the expression of these molecules in both liver and PB NK cells, indicating that these cells retain their ability to protect self-MHC class I-expressing cells from NK cell-mediated death. On the analyses of the cytotoxicity-associated receptors, including NKp30, NKp44, NKp46, and NKG2D, no statistically significant differences were found between PB and liver NK cells even after IL-2 stimulation (Fig. 4). Although liver NK cells tended to express higher levels of NKp44 and NKp46 than did PB NK cells, the differences did not reach statistical significance.

The proportions of CD56+CD3− NK and CD56+CD3+ NKT cells in LMNCs extracted from liver perfusates were significantly higher than those in PBMCs. a Flow cytometric (FCM) analyses of freshly isolated LMNCs obtained from liver perfusates and PBMCs from the corresponding donors were analyzed after staining with mAbs against CD3 and CD56. Lymphocytes were gated by forward scatter and side scatter. FCM profiles are representative of six and five independent experiments (using PBMCs and LMNCs, respectively). a CD3−CD56bright NK cell subset, b CD3−CD56dim NK cell subset, c CD3+CD56+ (NKT) cell subset. b Percentages of CD3−CD56bright NK, CD3−CD56dim NK, and CD3+CD56+ NKT cells among mononuclear cells (mean ± SEM, PBMCs; n = 6, LMNCs; n = 5). c Histograms showing the log fluorescence intensities obtained for TRAIL staining after gating CD3−CD56bright NK and CD3−CD56dim NK cell subsets of LMNCs and PBMCs from the corresponding donors. Data are shown as overlays with comparison against isotype controls. Histogram profiles are representative of independent experiments (PBMCs; n = 6, LMNCs; n = 5). d Mean fluorescence intensities (MFI) of TRAIL staining on NK cells freshly isolated from LMNCs and PBMCs (CD56bright NK cells; open column, CD56dim NK cells; closed column). Data represent mean ± SEM (PBMCs; n = 6 LMNCs; n = 5). Statistical analyses were performed using the paired Student’s t test (*P < 0.05). NK natural killer, TRAIL TNF-related apoptosis-inducing ligand, LMNC liver mononuclear cells, PBMC peripheral blood mononuclear cells, mAb monoclonal antibody

Phenotypic analyses of human PB and liver NK cells. a Flow cytometric (FCM) analyses of PBMCs and LMNCs cultivated with IL-2 for 5 days were performed after staining with mAbs against CD3 and CD56. FCM profiles are representative of five and six independent experiments (using PBMCs and LMNCs, respectively). Percentages of NK and NKT cells are indicated (mean ± SEM, PBMC; n = 5, LMNC; n = 6). b Expression of cytotoxic effector molecules on NK cell subsets among LMNCs or PBMCs freshly isolated or cultivated with or without IL-2 was analyzed. Expression of TRAIL and FasL on electronically gated CD3−CD56+ NK cells was analyzed by FCM. Numbers above the lesion marker line indicate the percentages of cells expressing TRAIL and FasL, and numbers below the line indicate the median florescence intensity of expression of whole NK cells (mean ± SEM, n = 4 each). PBMCs were obtained from the corresponding LMNC donor. Histogram profiles are representative of independent experiments. Dotted lines represent negative control staining with isotype-matched mAbs. *P < 0.05 PB NK cells versus liver NK cells

IL-2 stimulation maintained the expression of C-type lectin-like receptors and killer cell immunoglobulin-like receptors (KIR) in both human PB and liver NK cells. Expression of the C-type lectin-like receptors CD94, NKG2A and NKG2C, and KIR such as CD158a and CD158b on NK cell subsets among LMNCs or PBMCs freshly isolated or cultivated with or without IL-2 was analyzed. Expression on electronically gated CD3−CD56+ NK cells was analyzed by FCM. Numbers above the lesion marker line indicate the percentages of cells expressing each molecule, and numbers below the line indicate the median florescence intensity of expression of whole NK cells (mean ± SEM, n = 4 each). PBMCs were obtained from the corresponding LMNC donor. Histogram profiles are representative of independent experiments. Dotted lines represent negative control staining with isotype-matched mAbs. *P < 0.05 PB NK cells versus liver NK cells

IL-2 stimulation led to elevated expression of NKp30, NKp44, and NKG2D in both human PB and liver NK cells. Expression of the cytotoxicity-associated receptors, including NKp30, NKp44, NKp46, and NKG2D, in NK cell subsets among LMNCs or PBMCs freshly isolated or cultivated with or without IL-2 was analyzed. Expression in electronically gated CD3−CD56+ NK cells was analyzed by FCM. Numbers above the lesion marker line indicate the percentages of cells expressing each molecule, and numbers below the line indicate the median florescence intensity of expression of whole NK cells (mean ± SEM, n = 4 each). PBMCs were obtained from the corresponding LMNC donor. Histogram profiles are representative of independent experiments. Dotted lines represent negative control staining with isotype-matched mAbs.
Breast cancer cells express the death-inducing receptor
Susceptibility to TRAIL-induced apoptosis may be related to the expression levels of multiple receptors on target cells. TRAIL binds to at least four receptors: two of these are death-inducing receptors (TRAIL-R1/DR4 and TRAIL-R2/DR5) containing cytoplasmic death domains and signal apoptosis, whereas the other two are death-inhibitory receptors (TRAIL-R3/DcR1 and TRAIL-R4/DcR2) that lack a functional death domain and do not mediate apoptosis; all have similar affinities for TRAIL and the latter two may act as decoys [16, 17]. The susceptibility to TRAIL-induced apoptosis is related to the expression levels of those receptors in tumor cells. We investigated the expression patterns of TRAIL-DR and TRAIL-DcR in both normal mammary gland and breast cancer tissue samples. Ductal cells in normal mammary gland tissues expressed TRAIL-DR4 together with TRAIL-DcR1 (Fig. 5a). Breast cancer cells showed a much higher expression of TRAIL-DR4 than did normal mammary gland cells, but little TRAIL-DcR1, regardless of the HER2 type. Similar to the clinical breast cancer tissues, all the tested breast cancer cell lines expressed high TRAIL-DR4 together with TRAIL-DR5, but no TRAIL-DcR1 and TRAIL-DcR2, regardless of their HER2 status (Fig. 5b).

Breast cancer cells express death-inducing TRAIL-DR4 but lack death-inhibitory TRAIL-DcR1 and TRAIL-DcR2. a Immunohistochemical expression of TRAIL-DcR1, TRAIL-DcR2, TRAIL-DR4, and TRAIL-DR5 in normal breast tissue and tumor sites of HER2 (+)- and HER2 (−)-type breast cancer tissues. Immunopathological findings are representative of three individual samples in each breast cancer category. Magnification ×200. Scale bar 100 μm. b Surface expression of HER2 and TRAIL receptors on the surface of MDA-MB231, MDA-MB453, MDA-MB468, and MCF-7 was analyzed by FCM. Dotted lines represent negative control staining with isotype-matched mAbs. Numbers indicate the mean fluorescence intensity (MFI) of cells that stained positively for HER2 and TRAIL receptors. TRAIL, TNF-related apoptosis-inducing ligand; FCM flow cytometric, mAb monoclonal antibody, TNF tumor necrosis factor
IL-2-stimulated NK cells showed significant cytotoxicity against breast cancer cells
Cytotoxicity assays of NK cells isolated from LMNCs and PBMCs as effectors and various breast cancer cell lines as targets were performed. Cells were stimulated by 5-day culture with IL-2 before use in the cytotoxicity assays. Liver NK cells showed more vigorous cytotoxicity against all tested cell lines (MDA-MB231, MDA-MB453, MDA-MB468, and MCF-7) compared with PB NK cells (Fig. 6). Addition of trastuzumab enhanced the cytotoxicity of both liver and PB NK cells toward MDA-MB231, MDA-MB453, and MCF-7, which express HER2. Although MDA-MB468 has been reported to be a triple-negative breast cancer cell line [12], it seemed to express dim HER2 on phenotypic analysis of the breast cancer cell lines in this study (Fig. 5b). This may explain why trastuzumab did not promote the cytotoxicity of PB NK cells but somewhat enhanced the cytotoxicity of liver NK cells toward MDA-MB468. Nevertheless, these observations suggest the involvement of HER2/trastuzumab-mediated ADCC. Despite the strong cytotoxicity exhibited by IL-2-stimulated donor liver NK cells, their cytotoxicities toward one-haplotype identical allogeneic and autologous lymphoblasts were negligible (data not shown).

Liver NK cells showed more vigorous cytotoxicity against breast cancer cell lines compared with PB NK cells. Cytotoxic activities of NK cells isolated from IL-2-stimulated PBMCs and LMNCs with or without trastuzumab against target cells (MDA-MB231, MDA-MB453, MDA-MB468, and MCF-7) were analyzed by the 51Cr release assay. NK cells were isolated from PBMCs and LMNCs after stimulation with IL-2 for 5 days by magnetic sorting (purity > 90%). Data represent the mean ± SEM of values from triplicate samples and represent four similar experiments
TRAIL and perforin are involved in the cytotoxicity of NK cells against breast cancer cells
To determine the contribution of TRAIL to the cytotoxicity of NK cells against breast cancer cells, the effect of a neutralizing anti-TRAIL mAb was examined in a cytotoxicity assay with PB and liver NK cells as effectors and MDA-MB231 cells as the target. Both PB and liver NK cell-induced cytotoxicity was inhibited partially by the anti-TRAIL mAb alone and more profoundly by the combination of the anti-TRAIL mAb and CMA, indicating that TRAIL and perforin are involved in NK cell-mediated cytotoxicity (Fig. 7). Remarkable levels of inhibition of NK cell-induced cytotoxicity were observed with anti-TRAIL mAb at 18-h culture when compared with those at 4 h. This finding is consistent with the results of the previous study with a mouse model demonstrating that death receptor-mediated NK cell kill needs longer incubation times than perforin-mediated NK cell kill [18].

NK cell-induced cytotoxicity was inhibited partially by the anti-TRAIL mAb alone and more profoundly by the combination of anti-TRAIL mAb and CMA. Isolated PB and liver NK cell populations were used as effector cells (E) in assays of cytotoxicity against the target (T) MDA-MB231 human breast cancer cell line. Cytotoxicity assays were performed at an E/T ratio of 10:1, 5:1, or 2.5:1 in the presence or in the absence of anti-TRAIL (N2B2) mAb (10 μg/ml) and/or concanamycin A (CMA) (50 nmol/l). Data are the average ± SEM values from triplicate samples and represent four similar experiments, in which five different donor individuals were used (the results of four other experiments are shown in Supplementary Figures 1 and 2). Error bars not shown appear within the data point. NK natural killer, TRAIL tumor necrosis factor-related apoptosis-inducing ligand, mAb monoclonal antibody, FasL Fas ligand, E/T effector-to-target, CMA concanamycin A
NK cells were preferentially drawn by chemokines secreted from breast cancer cells, presumably through the CXCL10/CXCR3 axis
The distribution of NK cells is known to be associated with their expression of receptors and ligands for chemokines secreted from infectious or neoplastic sites [19–21]. We found that NK cells freshly isolated from PBMCs and LMNCs highly expressed CXCR3, which binds to the chemokines CXCL9, CXCL10, and CXCL11 secreted by breast cancer cells. IL-2 activation increased the levels of CXCR3 expression on both NK cell types (Fig. 8a, b). We further investigated the secretion activities of these various chemokines from the breast cancer cell lines. Significant levels of CXCL10 were detected in the culture supernatants of three of four breast cancer cell lines: MDA-MB231, MDA-MB453, and MDA-MB468 (Fig. 9a). PB or liver NK cells activated with IL-2 were cultured in the upper compartment of transwell tissue culture plates in the presence or in the absence of MDA-MB231 or MDA-MB468 cells in the lower compartment for 2 h. The migration of PB and liver NK cells through the membrane was markedly promoted by the presence of tumor cells in the lower compartment, suggesting that NK cells are preferentially drawn by chemokines secreted from tumor cells (Fig. 9b). Significant levels of CXCL10 were detected only in the culture supernatants in the lower compartment with MDA-MB231 (Fig. 9c), suggesting that the CXCL10/CXCR3 axis plays an important role in the accumulation of NK cells in tumor sites. The MDA-MB231 cell line produced a lot more CXCL10 when PB NK cells were added (Fig. 9a, c), suggesting that soluble factors secreted from NK cells promoted the production of CXCL10 from this cell line. Taken together with the fact that CXCL10 is an IFN-γ-inducible protein [22] and that IL-2 augments the active production of IFN-γ from NK cells, we could assume that IFN-γ secreted from NK cells promotes CXCL10 production from the breast cancer cell line, which in turn accelerates the migration of CXCR3-expressing NK cells into the tumor site. Consistent with this hypothesis, the CXCL10 levels were well correlated with IFN-γ levels in the culture supernatants of the cell migration assay (Fig. 9d). In addition, we directly confirmed that IFN-γ promoted the production of CXCL10 from MDA-MB231 and MDA-MB468 in a dose-dependent manner (Fig. 9e, f).

CXC chemokine receptor 3 (CXCR3) expression was significantly upregulated on both liver and PB NK cells after cultivation with IL-2. a Histograms representing the log fluorescence intensities for CXCR3 expression on freshly isolated and IL-2-stimulated liver and PB NK cells. Dotted lines represent negative control staining with isotype-matched mAbs. Numbers (mean ± SEM) indicate the percentages of cells in each group that were positive for CXCR3 expression (PBMCs; n = 6, LMNCs; n = 7). Histogram profiles are representative of independent experiments. b Numbers indicate the mean fluorescence intensity (MFI) of cells that stained positively for CXCR3 on liver and PB NK cells (fresh NK cells open column, activated NK cells closed column). Data represent mean ± SEM (PBMCs; n = 6, LMNCs; n = 7). Statistical analyses were performed using the paired Student’s t test (**P < 0.01). NK natural killer, LMNC liver mononuclear cell, PBMC peripheral blood mononuclear cell, mAb monoclonal antibody

NK cells were preferentially drawn by chemokines secreted from breast cancer cells, presumably through the CXCL10/CXCR3 axis. a Levels of various chemokines (CXCL9, CXCL10, and CXCL11) in the culture supernatants of breast cancer cell lines (MDA-MB231, MDA-MB453, MDA-MB468, and MCF-7) were analyzed using CBA Flex Sets. Supernatants were collected after 2 days of cultivation. b Migration assays were performed in transwell culture inserts with 3-μm pore filters. MDA-MB231 and MDA-MB468 cell lines were cultured in the lower chamber of the plate for 2 days and IL-2-activated NK cells from PBMCs and LMNCs were added to the upper chamber. After 2 h, the migrated NK cells were counted. Results are presented as mean migrated cell numbers ± SEM (n = 3). c Levels of chemokine in the medium of lower chambers in the migration assays described above were measured by CBA assay. Results are the average ± SEM values from triplicate samples and represent three individual experiments. d Levels of IFN-γ in the medium of lower chambers in the migration assays described above were measured by ELISA. The results are the average ± SEM values from triplicate samples and represent three individual experiments. e MDA-MB231 cells were cultured with various doses of IFN-γ for 2 and 6 h, and the levels of CXCL10 in the medium were measured by CBA assay. Results are the average ± SEM values from triplicate samples and represent three individual experiments. f MDA-MB468 cells were cultured with various doses of IFN-γ for 2 and 6 h, and the levels of CXCL10 in the medium were measured by CBA assay. Results are the average ± SEM values from triplicate samples and represent three individual experiments
Discussion
Human NK cells can be divided into the CD56bright and CD56dim subsets. These subsets have different phenotypic expression and may have different functions, although the direct functional significance of the expression levels of the CD56 antigen remains unknown. We previously demonstrated that CD56bright NK cells, which constitutively express low levels of TRAIL, are abundant in the liver [10]. CD56bright NK cells also constitutively express the high-affinity heterotrimeric IL-2R (IL-2Rαβγ) [23, 24]; hence, this subset has a high proliferative response to IL-2 and expand and survive through the upregulation of bcl-2 in vitro in response to IL-2 [25, 26]. In contrast, resting CD56dim NK cells, which express IL-2Rβγ only, show almost no proliferation in response to even high doses of IL-2 in vitro [23, 26]. In this study, CD56bright NK cells exclusively survived and significantly upregulated TRAIL expression after in vitro cultivation of both PBMCs and LMNCs with IL-2 (Fig. 2). IL-2 stimulation also increased the surface expression of inhibitory receptors such as the KIR, including CD158a/158b and C-type lectin-like receptors (the CD94/NKG2 complex). CD94, which is expressed on essentially all NK cells, uses HLA-E expression as a sensor for the overall HLA class I level of a cell. In contrast, individual KIR family members are expressed on certain NK cell subsets and exhibit finer specificity for HLA class I allotypes and can distinguish between groups of HLA-A, HLA-B, and HLA-C allotypes. Ligation of such KIRs/CD94 to HLA class I molecules on self cells results in inhibition of NK cell cytotoxic activity, as originally predicted by the “missing-self” hypothesis [1, 27]. This regulation ensures that cells expressing none, altered, or reduced MHC-I molecules, such as malignant or virus-infected cells, are eliminated by NK cells. The modulated expression of KIRs/CD94 by IL-2 is likely associated with the changed cytotoxic target-discriminating ability of NK cells upon their exposure to IL-2.
The significantly upregulated TRAIL expression on the IL-2-stimulated NK cells implies that they have the ability to target cancer cells expressing death-inducing receptors. TRAIL is a member of the TNF superfamily, which includes TNF and FasL [28]. The expression of TNF and FasL leads to damage of normal tissues in addition to their proapoptotic effect on transformed cells [29, 30], limiting their clinical applications. Conversely, TRAIL selectively induces apoptosis in transformed cells but not in most normal cells [28, 31, 32], making it a promising candidate for tumor therapy. However, intravenous delivery of recombinant TRAIL has met with problems, including a short pharmacokinetic half-life [32], necessitating frequent and high doses to produce the desired effect. The use of TRAIL-expressing NK cells as a delivery vector might promise both targeted and prolonged delivery of this death ligand.
TRAIL binds DR4 and DR5, leading to the formation of the death-inducing signaling complex and the Fas-associated protein with death domain. In turn, these complexes recruit caspase-8 (or caspase-10), which plays an important role in apoptosis induction either by direct activation of downstream effector caspases (caspase-3, caspase-6, and caspase-7) or by cleaving apoptotic molecules (Bcl-2 and Bcl-xL), resulting in further activation of the caspase-9 complex [33]. In this study, breast cancer cells of clinical samples showed much higher expression of TRAIL-DR4 than normal mammary glands but exhibited little TRAIL-DcR1, regardless of HER2 type. Similarly, all the tested breast cancer cell lines expressed TRAIL-DR4 but not TRAIL-DcR1 and TRAIL-DcR2, regardless of their HER2-status, suggesting that they are susceptible to TRAIL-induced apoptosis.
We tested various breast cancer cell lines to evaluate their susceptibility to NK cell-mediated cytotoxicity. Notably, liver NK cells showed more vigorous cytotoxicity against all the tested cell lines than did PB NK cells (Fig. 6), although the underlying mechanism remains unclear. The contribution of TRAIL to NK cell cytotoxicity was determined using the neutralizing anti-TRAIL mAb (Fig. 7). Trastuzumab addition remarkably enhanced the cytotoxicity of both NK cell types toward HER2-overexpressing breast cancer cell lines, indicating that HER2/trastuzumab-mediated ADCC was involved. As ADCC requires the activation and engagement of the CD16 FcγR on NK cells by Ab-coated targets, CD56dim NK cells, which highly express CD16 (Fcγ receptor III), are generally thought to exhibit greater levels of ADCC than do the CD56bright subset [34]. On the other hand, the majority of CD56bright NK cells expanded after activation with IL-2 expressed CD16 and efficiently mediated ADCC [20], explaining the HER2/trastuzumab-mediated ADCC observed in this study.
The cytotoxic ability of NK cells against cancer cells presumably requires contact between NK cells and their target cells. In general, NK cells are detected infrequently in tumors and their presence in the infiltrate consistently correlates with a good prognosis and increased patient survival [35, 36]. Chemokines acting on CXCR3 and CX3CR1 are considered major determinants of NK cell infiltration. CX3CR1 expression in gastric adenocarcinoma samples directly correlates with the number of NK cells infiltrating the tumor, and patients with higher CX3CL1 levels had a significantly better prognosis than patients with low CX3CL1 levels [37]. Similarly, our in vitro demonstration that the CXCL10/CXCR3 axis plays a role in the attraction between activated NK cells and breast cancer cells suggests that this chemokine system recruits NK cells to cancer cell sites and elicits antitumoral responses. In addition, we proposed a novel mechanistic paradigm in which IFN-γ secreted from NK cells promotes the production of CXCL10 from breast cancer cells, which in turn further accelerates the migration of CXCR3-expressing NK cells into the tumor site (Fig. 10).

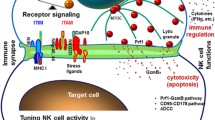
Mechanistic paradigm of interaction between NK and breast cancer cells. IFN-γ secreted from NK cells promotes the production of CXCL10 from breast cancer cells, which in turn further accelerates migration of CXCR3-expressing NK cells into the tumor site. Migrated NK cells kill breast cancer cells by either of the two major mechanisms that require direct contact between NK cells and target cells. In the first, cytoplasmic granule toxins, perforin, and granzymes are secreted by exocytosis and together induce apoptosis of the target cell. The second mechanism involves the engagement of death receptors on target cells by expressing of their cognate ligands (TRAIL) on NK cells, resulting in apoptosis of the target cells
Given the efficacy of NK cells to selectively eliminate abnormal cells, a variety of approaches have been taken to selectively augment NK cell response to tumors [38, 39]. Several therapeutic cytokines primarily act through NK cells (e.g., IL-2, IL-12, IL-15, and IFNs), and many studies have shown that activation of NK cell differentiation and function leads to more efficient elimination of tumor growth 9 [40–44]. Despite these promising advances, the systemic administration of cytokines, such as IL-2, which non-specifically activate a broad range of immune cell types, is associated with significant toxicity [40, 45]. Recent animal experiments have demonstrated the ability of adoptive transfer of NK cells to mount a therapeutic antitumor response [46, 47], and translational clinical research suggests that NK cells are useful for controlling human malignancy [48–50]. Our results have proven that PB NK cells can kill breast cancer cells and liver NK cells can hinder metastasis of breast cancer to the liver, which suggests the potential therapeutic use of NK cells, i.e., by either activation of endogenous NK cells or adoptive transfer of in vitro-activated autologous NK cells. Although liver NK cells displayed higher cytotoxicity than PB NK cells, no clinically applicable method for obtaining liver NK cells from patients with breast cancer has yet been established. Alternatively, locally infusing IL-2 into the liver through the portal vein likely activates endogenous liver NK cells, which in turn might infiltrate or accumulate to the tumor site probably through the CXCL10/CXCR3 axis.
Abbreviations
- NK:
-
Natural killer
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- PBMC:
-
Peripheral blood mononuclear cell
- LMNC:
-
Liver mononuclear cell
- mAbs:
-
Monoclonal antibodies
References
Trinchieri G (1989) Biology of natural killer cells. Adv Immunol 47:187–376
Robertson MJ, Ritz J (1990) Biology and clinical relevance of human natural killer cells. Blood 76(12):2421–2438
Cullen SP, Martin SJ (2008) Mechanisms of granule-dependent killing. Cell Death Differ 15(2):251–262
Pardo J, Aguilo JI, Anel A, Martin P, Joeckel L, Borner C et al (2009) The biology of cytotoxic cell granule exocytosis pathway: granzymes have evolved to induce cell death and inflammation. Microbes Infect 11(4):452–459
Sjostrom-Mattson J, Von Boguslawski K, Bengtsson NO, Mjaaland I, Salmenkivi K, Blomqvist C (2009) The expression of p53, bcl-2, bax, fas and fasL in the primary tumour and lymph node metastases of breast cancer. Acta Oncol 48(8):1137–1143
Lahiry L, Saha B, Chakraborty J, Adhikary A, Hossain DM et al (2010) Theaflavins target Fas/caspase-8 and Akt/pBad pathways to induce apoptosis in p53-mutated human breast cancer cells. Carcinogenesis 31(2):259–268
Takahashi M, Ogasawara K, Takeda K, Hashimoto W, Sakihara H, Kumagai K et al (1996) LPS induces NK1.1+ alpha beta T cells with potent cytotoxicity in the liver of mice via production of IL-12 from Kupffer cells. J Immunol 156(7):2436–2442
Crispe IN, Mehal WZ (1996) Strange brew: T cells in the liver. Immunol Today 17(11):522–525
Seki S, Habu Y, Kawamura T, Takeda K, Dobashi H, Ohkawa T et al (2000) The liver as a crucial organ in the first line of host defense: the roles of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells in T helper 1 immune responses. Immunol Rev 174:35–46
Ishiyama K, Ohdan H, Mitsuta H, Arihiro K, Asahara T (2006) Difference in cytotoxicity against hepatocellular carcinoma between liver and periphery natural killer cells in humans. Hepatology 43(2):362–372
Ochi M, Ohdan H, Mitsuta H, Onoe T, Tokita D, Hara H et al (2004) Liver NK cells expressing TRAIL are toxic against self hepatocytes in mice. Hepatology 39(5):1321–1331
Rahman M, Davis SR, Pumphrey JG, Bao J, Nau MM, Meltzer PS et al (2009) TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res Treat 113(2):217–230
Dastjerdi K, Tabar GH, Dehghani H, Haghparast A (2011) Generation of an enriched pool of DNA aptamers for an HER2-overexpressing cell line selected by cell SELEX. Biotechnol Appl Biochem 58(4):226–230
Kataoka T, Shinohara N, Takayama H, Takaku K, Kondo S, Yonehara S et al (1996) Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol 156(10):3678–3686
Kawarabayashi N, Seki S, Hatsuse K, Ohkawa T, Koike Y, Aihara T et al (2000) Decrease of CD56(+)T cells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibility to hepatocellular carcinoma. Hepatology 32(5):962–969
Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J et al (1997) The receptor for the cytotoxic ligand TRAIL. Science 276(5309):111–113
Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D et al (1997) Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277(5327):818–821
Wallin RP, Screpanti V, Michaelsson J, Grandien A, Ljunggren HG (2003) Regulation of perforin-independent NK cell-mediated cytotoxicity. Eur J Immunol 33(10):2727–2735
Salazar-Mather TP, Orange JS, Biron CA (1998) Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)-dependent pathways. J Exp Med 187(1):1–14
Guimond MJ, Wang B, Croy BA (1998) Engraftment of bone marrow from severe combined immunodeficient (SCID) mice reverses the reproductive deficits in natural killer cell-deficient tg epsilon 26 mice. J Exp Med 187(2):217–223
Hedrick JA, Saylor V, Figueroa D, Mizoue L, Xu Y, Menon S et al (1997) Lymphotactin is produced by NK cells and attracts both NK cells and T cells in vivo. J Immunol 158(4):1533–1540
Groom JR, Luster AD (2011) CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89(2):207–215
Caligiuri MA, Zmuidzinas A, Manley TJ, Levine H, Smith KA, Ritz J (1990) Functional consequences of interleukin 2 receptor expression on resting human lymphocytes. Identification of a novel natural killer cell subset with high affinity receptors. J Exp Med 171(5):1509–1526
Nagler A, Lanier LL, Phillips JH (1990) Constitutive expression of high affinity interleukin 2 receptors on human CD16-natural killer cells in vivo. J Exp Med 171(5):1527–1533
Caligiuri MA, Murray C, Robertson MJ, Wang E, Cochran K, Cameron C et al (1993) Selective modulation of human natural killer cells in vivo after prolonged infusion of low dose recombinant interleukin 2. J Clin Invest 91(1):123–132
Baume DM, Robertson MJ, Levine H, Manley TJ, Schow PW, Ritz J (1992) Differential responses to interleukin 2 define functionally distinct subsets of human natural killer cells. Eur J Immunol 22(1):1–6
Karre K, Ljunggren HG, Piontek G, Kiessling R (1986) Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319(6055):675–678
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK et al (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3(6):673–682
Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y et al (1993) Lethal effect of the anti-Fas antibody in mice. Nature 364(6440):806–809
Nagata S (1997) Apoptosis by death factor. Cell 88(3):355–365
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA et al (1999) Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 104(2):155–162
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith T, Kubin M et al (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5(2):157–163
Herr I, Debatin KM (2001) Cellular stress response and apoptosis in cancer therapy. Blood 98(9):2603–2614
Nagler A, Lanier LL, Cwirla S, Phillips JH (1989) Comparative studies of human FcRIII-positive and negative natural killer cells. J Immunol 143(10):3183–3191
Ishigami S, Natsugome S, Tokuda K, Nakajo A, Xiangming Che, Iwashige H et al (2000) Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 88(3):577–583
Villegas FR, Santiago C, Villarrubia VG, Jimenez R, Chillon MJ, Jareno J et al (2002) Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 35(1):23–28
Hyakudomi M, Matsubara T, Hyakudomi R, Yamamoto T, Kinugasa S, Yamanoi A et al (2008) Increased expression of fractalkine is correlated with a better prognosis and an increased number of both CD8+ T cells and natural killer cells in gastric adenocarcinoma. Ann Surg Oncol 15(6):1775–1782
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK et al (2005) Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105(8):3051–3057
Klingemann HG, Martinson J (2004) Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy 6(1):15–22
Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S et al (1987) A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med 316(15):889–897
Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M et al (1993) Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 178(4):1223–1230
Takeda K, Hayakawa Y, Smyth ML, Kayagaki N, Yamaguchi N, Kakuta S et al (2001) Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 1:94–100
Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M et al (2000) Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med 191(5):771–780
Smyth MJ, Cretney E, Takeda K, Wiltrout RH, Sedger LM, Kayagaki N et al (2001) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med 193(6):661–670
Talmadge JE, Phillips H, Schindler J, Tribble H, Pennigton R (1987) Systematic preclinical study on the therapeutic properties of recombinant human interleukin 2 for the treatment of metastatic disease. Cancer Res 47(21):5725–5732
Helms MW, Prescher JA, Cao YA, Schaffert S, Contag CH (2010) IL-12 enhances efficacy and shortens enrichment time in cytokine-induced killer cell immunotherapy. Cancer Immunol Immunother 59(9):1325–1334
Pegram HJ, Haynes NM, Smyth MJ, Kershaw MH, Darcy PK (2010) Characterizing the anti-tumor function of adoptively transferred NK cells in vivo. Cancer Immunol Immunother 59(8):1235–1246
Iliopoulou EG, Kountourakis P, Karamouzis MV, Doufexis D, Ardavanis A, Baxevanis CN et al (2010) A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol Immunother 59(12):1781–1789
Milani V, Stangl S, Issels R, Gehrmann M, Wagner B, Hube K (2009) Anti-tumor activity of patient-derived NK cells after cell-based immunotherapy—a case report. J Transl Med 7:50
Knorr DA, Kaufman DS (2010) Pluripotent stem cell-derived natural killer cells for cancer therapy. Transl Res 156(3):147–154
Acknowledgments
We thank Drs. Kohei Ishiyama, and Masahiro Ohira for their advice and encouragement and Drs. Doskali Marlen, Yuka Igarashi and Nabin Basnet, and Ms. Yuko Ishida and Ms. Midori Kiyokawa for their expert technical assistance. This work was supported by a Grant-in-Aid for Scientific Research (A) from the Japan Society for the Promotion of Science and a Grant-in-Aid for the Research on Hepatitis and BSE from the Japanese Ministry of Health, Labour and Welfare.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. 1
The results of 4 other similar experiments using liver NK cells to that shown in Fig. 7. Isolated liver NK cell populations were used as effector cells (E) in assays of cytotoxicity against the target (T) MDA-MB231 human breast cancer cell line. Cytotoxicity assays were performed at an E/T ratio of 10:1, 5:1, or 2.5:1 in the presence or absence of anti-TRAIL (N2B2) mAb (10 μg/mL) and/or concanamycin A (CMA) (50 nmol/L). Data are the average ± SEM values from triplicate sample. Error bars not shown appear within the data point. (TIFF 103 kb)
Fig. 2
The results of 4 other similar experiments using PB NK cells to that shown in Fig. 7. Isolated PB NK cell populations were used as effector cells (E) in assays of cytotoxicity against the target (T) MDA-MB231 human breast cancer cell line. Cytotoxicity assays were performed at an E/T ratio of 10:1, 5:1, or 2.5:1 in the presence or absence of anti-TRAIL (N2B2) mAb (10 μg/mL) and/or concanamycin A (CMA) (50 nmol/L). Data are the average ± SEM values from triplicate sample. Error bars not shown appear within the data point. (TIFF 102 kb)
Rights and permissions
About this article
Cite this article
Kajitani, K., Tanaka, Y., Arihiro, K. et al. Mechanistic analysis of the antitumor efficacy of human natural killer cells against breast cancer cells. Breast Cancer Res Treat 134, 139–155 (2012). https://doi.org/10.1007/s10549-011-1944-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-011-1944-x