
Abstract
In order to adequately evaluate the clinical relevance of genetic testing in sporadic breast and ovarian cancer patients, we offered comprehensive BRCA1/2 mutation analysis in patients without a family history for the disease. We evaluated the complete coding and splice site regions of BRCA1/2 in 193 sporadic patients. In addition, a de novo mutation was further investigated with ultra deep sequencing and microsatellite marker analysis. In 17 patients (8.8%), a deleterious germline BRCA1/2 mutation was identified. The highest mutation detection ratio (3/7 = 42.9%) was obtained in sporadic patients diagnosed with breast and ovarian cancer after the age of 40. In 21 bilateral breast cancer patients, two mutations were identified (9.5%). Furthermore, 140 sporadic patients with unilateral breast cancer were investigated. Mutations were only identified in patients diagnosed with breast cancer before the age of 40 (12/128 = 9.4% vs. 0/12 with Dx > 40). No mutations were detected in 17 sporadic male breast cancer and 6 ovarian cancer patients. BRCA1 c.3494_3495delTT was identified in a patient diagnosed with breast and ovarian cancer at the age of 52 and 53, respectively, and was proven to have occurred de novo at the paternal allele. Our study shows that the mutation detection probability in specific patient subsets can be significant, therefore mutation analysis should be considered in sporadic patients. As a consequence, a family history for the disease and an early age of onset should not be used as the only criteria for mutation analysis of BRCA1/2. The relatively high mutation detection ratio suggests that the prevalence of BRCA1/2 may be underestimated, especially in sporadic patients who developed breast and ovarian cancer. In addition, although rare, the possibility of a de novo occurrence in a sporadic patient should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Germline BRCA1 (MIM 113705) [1] and BRCA2 (MIM 600185) [2, 3] mutations confer high risks for breast and ovarian cancer and are most prevalent in patients with a family history for the disease. The incidence of mutations in high-risk families varies widely among different populations; some present a wide spectrum of different mutations, while in particular ethnic groups specific mutations show a high frequency due to a founder effect. For example, approximately 2.5% of all people of Ashkenazi Jewish descent carry one of three ancient mutations in BRCA1 or BRCA2 (c.68_69delAG (185delAG) and c.5266dupC (5382insC) in BRCA1 and c.5946delT (6174delT) in BRCA2) [4].
Fewer reports and data on the prevalence of BRCA1 and BRCA2 germline mutations in sporadic breast and ovarian cancer patients are available, since inclusion criteria for BRCA mutation testing are most often based on the family history. A negative family history can be the result of a small family size, predominance of males in the family, or incomplete penetrance [5]. For sporadic patients, genetic testing is often limited to patients with early age at onset, bilaterality or tumor characteristics like multifocality, multicentricity, and triple-negative breast tumors, as these features increase the probability to detect a germline BRCA1 or 2 mutation.
Despite the high number of BRCA1/2 mutations identified, de novo BRCA mutations are rare. To the best of our knowledge, to date only two de novo mutations in BRCA1 are reported [6, 7] and five in BRCA2 [8–12]. All these mutations were identified in patients with tumors occurring before the age of 40.
In this study, we present our data on the prevalence of BRCA1&2 mutations in 193 sporadic Belgian breast and/or ovarian cancer patients. Furthermore, we present the identification of a de novo BRCA1 mutation in a patient diagnosed with breast cancer and ovarian cancer in her fifties.
Materials and methods
Patient population
The patients evaluated in this study are a subgroup of all Belgian patients referred to our center for diagnostic testing for BRCA1 and BRCA2. All the tested individuals provided a signed informed consent after appropriate genetic counseling. Personal and family histories of all patients were recorded, including ages of diagnosis of all cancers.
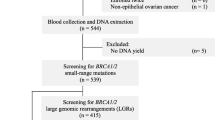
We investigated 193 breast and/or ovarian cancer patients without any family history for the disease. The majority of the study population (128 patients) are unilateral breast cancer patients who were selected because of an early age of onset (<40 years). Additionally, we analyzed 12 unilateral breast cancer patients with a later age at onset (Dx > 40). Furthermore, we investigated the BRCA1/2 coding region in 21 bilateral breast cancer patients and six ovarian cancer patients with an age at diagnosis <50. Male breast cancer patients (n = 17) and patients who developed breast in combination with ovarian cancer (n = 9) were selected without limitations based on the age of onset.
Mutation detection in BRCA1/2
Genomic DNA was isolated from peripheral blood mononuclear cells of all patients. The complete coding sequence and flanking splice site regions of BRCA1/2 were evaluated, by a wide variety of techniques, since these patients were selected retrospectively. Analysis of the samples was performed with a combination of techniques: Protein Truncation Test (PTT) [13], Denaturing Gradient Gel Electroforesis (DGGE) [14], High Resolution Melting Curve Analysis (HRMCA) [15, 16], or Sanger sequencing.
In addition, large intragenic rearrangements in BRCA1 and BRCA2 were evaluated with Multiplex Ligation Probe Amplification (MLPA).
Paternity testing
We identified a BRCA1 mutation c.3494_3495delTT (p.Phe1165fs) in one of the patients, which was absent in both parents. Paternity was verified using Powerplex16 assay (Promega). The PowerPlex® 16 System allows the co-amplification and three-color detection of sixteen loci (fifteen STR loci and Amelogenin). The system contains the loci Penta E, D18S51, D21S11, TH01, D3S1358, FGA, TPOX, D8S1179, vWA, Amelogenin, Penta D, CSF1PO, D16S539, D7S820, D13S317, and D5S818. One primer specific for Penta E, D18S51, D21S11, TH01, and D3S1358 is labeled with fluorescein (FL); one primer specific for FGA, TPOX, D8S1179, vWA, and Amelogenin is labeled with carboxy-tetramethylrhodamine (TMR); and one primer specific for Penta D, CSF1PO, D16S539, D7S820, D13S317, and D5S818 is labeled with 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxy-fluorescein (JOE). All the 16 loci are amplified simultaneously in a single tube and analyzed on an ABI3730 (Applied Biosystems). Data are analyzed with the Genemapper v1 software. The power of exclusion of this system exceeds 0.999998 in all populations tested.
Microsatellite marker analysis
To determine if a possible de novo mutation (BRCA1 c.3494_3495delTT) originated on the maternal or paternal allele, the region encompassing the BRCA1 gene was investigated with microsatellite markers in the parents, sibs, and children of the proband. Twelve microsatellite markers within or flanking BRCA1 were selected, spanning a 3 cM region. A schematic overview of the used markers and their location is shown in Fig. 1. Primer sequences for amplification of these markers were obtained from the Genome database. The forward primer was labeled with a FAM label and the amplification products were separated with capillary electrophoresis on an ABI3730 (Applied Biosystems). Plots were analyzed with the Genemapper software.

Schematic overview of the location of the used microsatellite markers. Thirteen microsatellite markers within or flanking BRCA1 were selected, spanning a 3.4 Mb region
Deep sequencing
Mosaicism in the parents of the patient with the possible de novo mutation (BRCA1 c.3494_3495delTT (p.Phe1165fs) was further investigated with ultra deep sequencing. The relevant amplicon was sequenced on a GS-FLX instrument (454-Roche). Data analysis was performed with the commercial available software Nextgene v2. (Softgenetics) [17].
Statistical analysis
Comparison of mutation prevalence between groups was performed with the chi-square test. For the statistical analysis of the mutation prevalence of smaller subgroups (n < 40), the Fisher’s exact test was used.
Results and discussion
We investigated the complete coding region of BRCA1&2 in 193 breast and/or ovarian cancer patients, without evidence for a family history of the disease. These patients were selected because they presented early onset or bilateral or multifocal tumors. In 17 (8.8%), a deleterious germline BRCA1/2 mutation was identified (an overview is shown in Table 1).
In three patients, previously unreported mutations were identified: BRCA1 c.3494_3495delTT, BRCA2 c.6948_6949insTT, and BRCA1 c.2507_2508delAA. These were, therefore, considered as potentially de novo. For two of them parental DNA was available to verify this hypothesis.
For the patient with the BRCA2 c.6948_6949insTT mutation; Sanger sequencing showed that she inherited the mutation from her father. An autosomal dominant inheritance pattern for this mutation is masked by the predominance of males in this branch of the pedigree (father has only male siblings).
BRCA1 c.3494_3495delTT was identified in a patient diagnosed with breast and ovarian cancer at the age of 52 and 53, respectively. This mutation was detected with high resolution melting curve analysis in our routine diagnostic screening [16]. Sanger sequencing revealed a truncating BRCA1 mutation c.3494_3495delTT (p.Phe1165fs) which to the best of our knowledge, has not yet been described before (Fig. 2).

Difference plot and Sanger sequencing of BRCA1 c.3494_3495delTT (p.Phe1165fs). Aberrant melting curves by high resolution melting curve analysis for one of the amplicons in exon 11 were detected in our routine diagnostic screening. A frequent polymorphism BRCA1 c.3548 A>G was identified in most of the samples. Sanger sequencing revealed a truncating BRCA1 mutation c.3494_3495delTT (p.Phe1165fs) in the sample
The mutation was not detectable in DNA extracted from blood lymphocytes of both parents by Sanger sequencing. Since the lower limit to detect mosaicism with Sanger sequencing is around 15% [18], we investigated possible mosaicism in lymphocytes with ultra deep sequencing on the GS-FLX instrument (454/Roche). The relevant amplicon was deep sequenced for the patient DNA, the maternal and the paternal sample. This ultradeep sequencing allows the identification of low grade mosaic variants [19]. The mutation was detected in 43% of the reads in the patient at 157-fold coverage, but was absent in the maternal and paternal sample both covered 1,840 and 2,780 fold, respectively, consistent with a de novo occurrence. A prevalence of a variant in 43% of all reads is consistent with 50% heterozygosity as heterozygous mutations can be present in 25–60% of the reads [17]. The coverage of 1,840 obtained for the maternal sample allows detection of mosaic mutations present in 5% of the blood lymphocytes with a probability of ≥99%; the higher coverage of the paternal sample (2,780×), results in a lower detection limit of 2% mosaicism with a 99% probability. Deeper coverage would be needed to detect variants with a frequency lower than 2% with a 99% probability.
To determine if the mutation originated on the maternal or paternal allele, the region encompassing the BRCA1 gene was investigated with 12 microsatellite markers in the parents, sibs, and children of the proband. Fragment analysis revealed that the mutation originated on the paternal allele of the proband. Five of her sibs inherited the same paternal allele but do not carry the mutation, providing additional evidence for the de novo occurrence of this mutation (data are shown in Fig. 3).

Microsatellite marker investigation of the de novo mutation BRCA1 c.3494_3495delTT. To determine if the mutation originated on the maternal or paternal allele, the region encompassing the BRCA1 gene was investigated with microsatellite markers in the parents, sibs, and children of the proband. Fragment analysis revealed that the mutation originated on the paternal allele of the proband. Five of her sibs inherited the same paternal allele but do not carry the mutation, providing additional evidence of the de novo occurrence of this mutation
Our data suggest a de novo event in a testicular germ cell, although a zygotic origin cannot be excluded. This is in agreement with previous findings in de novo mutations in BRCA1 and BRCA2, in which the paternal origin could be ascertained [8, 12]. Of note, the age of the patients’ father was only 29 years at the time of birth of his daughter. It has been suggested that the origin of new mutations may be influenced by both genomic imprinting effects and the increased number of cell divisions in spermatogenesis compared with oogenesis. A mainly paternal origin of mutations is seen in other hereditary syndromes as well, for instance in FGFR3, FGFR2, and RET, de novo mutations have been analyzed and all shown to be of paternal origin [20–22].
Not all disorders show such an extraordinary paternal bias, some single gene traits show only a moderate paternal bias in the male:female mutation ratio and a small paternal age effect. An example is neurofibromatosis type 1. The NF1 gene has one of the highest mutation rates. Many of the mutations are intragenic deletions, which are more common in larger genes. Whereas base substitutions occur primarily in males and are age-dependent, small chromosomal changes (intragenic deletions) are not age-dependent because they occur by different mechanisms [23].
This is the first de novo BRCA1 mutation identified in a patient with age of onset for the disease after the age of 50. In all patients previously reported with de novo mutations, breast tumors occurred at young age (<40 years), which may reflect recruitment bias, since genetic testing in sporadic cases is most often limited to patients with early onset breast or ovarian cancer. To our knowledge, only two other de novo BRCA1 mutations have been reported to date. The prevalence of de novo BRCA1 and BRCA2 mutations is still unknown and making a reliable estimation is challenging. Taking into account that recurrent BRCA1/2 mutations might appear as de novo alterations, might increase the prevalence to a higher rate than generally accepted [12]. As we could not evaluate parental DNA for all mutations detected in our sporadic patient population, the prevalence we observed might be an underestimation.
The majority of the mutations identified in this study (10/17), are well known founder mutations in our population. Although the Belgian population has a mixed ethnicity, we do see a considerable number of founder mutations in our population [24, 25]. In our overall patient population, 58 distinct mutations were detected, 17 of those represent approximately 75% of the 206 BRCA1/2 mutations identified to date in our center. Ten of the 17 mutations detected in this study are part of this top 17. Three mutations (BRCA1 p.Ser868X, BRCA2 c.4936_4939del4, and BRCA2 c.469_470delAA) are less prevalent in the Belgian population, but are reported in literature as founder mutations in other populations (Table 1).
We detected BRCA1/2 mutations in 189 of 1,170 (17%) unrelated index patients with a family history for breast/ovarian cancer referred to our center for genetic testing. This is statistically significantly higher than in our sporadic group (17/193 = 8.8%) (P < 0.01), demonstrating the relevance of inclusion criteria based on a family history. However, some subgroups of sporadic patients investigated in this study revealed a much higher frequency (see below and Table 2), indicating the importance of genetic testing for these patients.
Mutation analysis in 140 patients with unilateral breast cancer led to the identification of 12 germline mutations (8.6%). However, all the 12 mutations were detected in the subgroup of 128 patients diagnosed before the age of 40 (9.4%), of which three in the group of 55 patients diagnosed with unilateral breast cancer between the age of 35 and 40. Therefore, we have no evidence for a contribution of inherited BRCA1/2 mutations in unilateral breast cancer patients diagnosed in their forties, however, this observation is based on the analysis of only 12 patients. In the subgroup of 30 patients with at least two BRCA1/2 associated tumors (bilateral breast cancer or breast and ovarian cancer), we identified five mutations (16.7%). Interestingly, mutations were more prevalent in the patients diagnosed with their first tumor after the age of 40 (4/16 = 25%) compared to the group diagnosed before 40 (1/14 = 7.1%), however, the difference is not statistically significant (P = 0.36). Especially in patients with both breast and ovarian cancer (n = 9), a high mutation detection ratio was obtained (3/9 = 30%), even with a diagnosis at a more advanced age. Therefore, testing these patients is mandatory, independent of the age of onset. An overview of the prevalence of BRCA1/2 mutations in the different subgroups of our study population is given in Table 2.
The average age to develop breast cancer caused by a germline mutation is calculated to be 42 years for BRCA1 and 49 years for BRCA2 [26]. In BRCA1 mutation carriers, ovarian cancer rarely occurs before the age of 40 years, while 50 years seem to be a critical age for ovarian cancer in BRCA2 mutation carriers [26, 27]. In only two of our sporadic patients diagnosed with breast cancer before the age of 40 a BRCA2 mutation was identified (2x BRCA2 c.6280_6281delTT), in all other cases mutations in BRCA1 were detected. This is in agreement with the study of Peto et al. [28], who concluded that BRCA1 mutations may be more penetrant at young ages compared to BRCA2 mutations. Average age at diagnosis for breast cancer patients in whom a BRCA1 mutation was identified in our study population was 35.8 years [range: 27–53] compared to 38.4 years [range: 32–45] for BRCA2 mutations. Young age at primary breast cancer diagnosis is associated with an increased susceptibility to bilateral breast cancer, arguably due to the remaining life span and the resulting interval at which the patient remains at risk for a second primary tumor. In total, we investigated 21 patients diagnosed with bilateral breast cancer. All of these cases were diagnosed with their first breast cancer before the age of 50, but in only two patients a pathogenic BRCA mutation was identified.
We investigated the complete BRCA1/2 coding sequence in six sporadic ovarian cancer patients. Five of them were diagnosed with ovarian cancer at a young age (range 25–35 years) but no BRCA1 or BRCA2 mutations were detected. The estimated prevalence of BRCA1 mutations in familial ovarian cancer patients is 5% [29]. Although genetic testing to these five patients was offered because of their very young age of onset, other genetic factors may have contributed to the tumor development.
We identified no BRCA1/2 mutations in 17 sporadic male breast cancer patients (mean age at diagnosis: 52, range 25–72). As male breast cancer is rare, genetic testing is offered to these patients since it is believed to result from genetic susceptibility. Population and clinic based studies estimate the prevalence of BRCA2 mutations in males from 4% up to 40% depending on the ethnic background of the studied population [30–34]. One extreme example is the Icelandic population where 40% of all male breast cancers diagnosed carry the BRCA2 999del5 (BRCA2 c.771_775del) mutation. This is mainly due to a founder effect and is very unlikely to be replicated in other populations. The absence of BRCA2 mutations in our male sporadic breast cancer patients might be due to small sample size since we detected a BRCA2 mutation in 14% of all males with a family history for the disease referred to our center for genetic testing.
A BRCA1/2 mutation occurrence of ~10% is in agreement with previously reported findings in young (age at diagnosis <45 years) sporadic breast cancer patients in different populations [28, 35, 36]. However, prevalence is population dependent; for instance, the prevalence of BRCA1/2 mutations in British sporadic patients is lower (<4%) [37].
The ratio of BRCA1 to BRCA2 mutations varies widely between subpopulations globally. In this study, the prevalence of BRCA1 mutations is higher compared to BRCA2 mutations: 70% (12/17) of the mutations detected in the sporadic patients are present in BRCA1, this is higher than the prevalence of BRCA1 mutations (115/189 = 61%) in patients with a family history of breast/ovarian cancer, however, the difference is not statistically significant (P < 0.2). This increased prevalence of BRCA1 mutations can be explained by the higher penetrance of BRCA1 mutations in patients mainly selected on early age of onset, but also to founder effects, since the two most prevalent mutations (c.2380dupG and c.212+3A>G) in our population are both BRCA1 mutations.
Several tools to predict the likelihood for a patient to carry a mutation have been developed, unfortunately for most of them risk estimation is based on a family history for the disease. Important and widely used examples are the Claus, Tyrer-Cuzick, and BRCAPRO model but these cannot be reliably used for patients without any family history for breast or ovarian cancer. Our data and other studies demonstrate that these women may have a significant risk to carry a BRCA1 or BRCA2 mutation. Suggestions to adopt or expand the inclusion criteria for sporadic breast cancer patients have already been made [38]. Kwon et al. [38] suggest offering genetic testing to all women with triple-negative breast cancers diagnosed before the age of 50. Our study shows that a cut-off based on the age of onset cannot be used as the sole selection criterion for genetic testing of sporadic breast/ovarian cancer patients.
Conclusion
Selection of breast cancer patients based on the family history will not allow detection of all BRCA1/2 carriers that may benefit from preventive interventions. However, formulating a general cost effective set of guidelines to identify all sporadic BRCA1/2 mutation carriers is challenging. Our study proves that BRCA1/2 mutation detection in sporadic patients is worthwhile, even with diagnoses at later age, especially in patients with two tumors. The identification of a de novo BRCA1 mutation in a sporadic patient diagnosed with breast and ovarian cancer in her fifties, suggests that the prevalence of BRCA1/2 de novo mutations may currently be underestimated.
References
Miki Y et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266(5182):66–71
Wooster R et al (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378(6559):789–792
Wooster R et al (1994) Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12–13. Science 265(5181):2088–2090
Kauff ND et al (2002) Incidence of non-founder BRCA1 and BRCA2 mutations in high risk Ashkenazi breast and ovarian cancer families. J Med Genet 39(8):611–614
Gayther SA et al (1997) Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat Genet 15(1):103–105
Tesoriero A et al (1999) De novo BRCA1 mutation in a patient with breast cancer and an inherited BRCA2 mutation. Am J Hum Genet 65(2):567–569
Edwards E et al (2009) Identification of a de novo BRCA1 mutation in a woman with early onset bilateral breast cancer. Fam Cancer 8(4):479–482
Diez O et al (2010) A novel de novo BRCA2 mutation of paternal origin identified in a Spanish woman with early onset bilateral breast cancer. Breast Cancer Res Treat 121(1):221–225
Hansen TV et al (2008) Novel de novo BRCA2 mutation in a patient with a family history of breast cancer. BMC Med Genet 9:58
Marshall M, Solomon S, Lawrence Wickerham D (2009) Case report: de novo BRCA2 gene mutation in a 35-year-old woman with breast cancer. Clin Genet 76(5):427–430
Robson M et al (2002) Unique de novo mutation of BRCA2 in a woman with early onset breast cancer. J Med Genet 39(2):126–128
van der Luijt RB et al (2001) De novo recurrent germline mutation of the BRCA2 gene in a patient with early onset breast cancer. J Med Genet 38(2):102–105
Claes K et al (1999) Mutation analysis of the BRCA1 and BRCA2 genes results in the identification of novel and recurrent mutations in 6/16 flemish families with breast and/or ovarian cancer but not in 12 sporadic patients with early-onset disease. Mutations in brief no. 224. Online. Hum Mutat 13(3):256
van der Hout AH et al (2006) A DGGE system for comprehensive mutation screening of BRCA1 and BRCA2: application in a Dutch cancer clinic setting. Hum Mutat 27(7):654–666
De Leeneer K et al (2009) Genotyping of frequent BRCA1/2 SNPs with unlabeled probes: a supplement to HRMCA mutation scanning, allowing the strong reduction of sequencing burden. J Mol Diagn JMD 11(5):415–419
De Leeneer K et al (2008) Rapid and sensitive detection of BRCA1/2 mutations in a diagnostic setting: comparison of two high-resolution melting platforms. Clin Chem 54(6):982–989
De Leeneer K et al (2011) Massive parallel amplicon sequencing of the breast cancer genes BRCA1 and BRCA2: opportunities, challenges, and limitations. Hum Mutat 32(3):335–344
Rohlin A et al (2009) Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum Mutat 30(6):1012–1020
Thomas RK et al (2006) Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med 12(7):852–855
Carlson KM et al (1994) Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet 55(6):1076–1082
Schuffenecker I et al (1997) Prevalence and parental origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma, Le Groupe d’Etude des Tumeurs a Calcitonine. Am J Hum Genet 60(1):233–237
Glaser RL et al (2000) Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. Am J Hum Genet 66(3):768–777
Lazaro C et al (1996) Sex differences in mutational rate and mutational mechanism in the NF1 gene in neurofibromatosis type 1 patients. Hum Genet 98(6):696–699
Claes K et al (1999) Mutation analysis of the BRCA1 and BRCA2 genes in the Belgian patient population and identification of a Belgian founder mutation BRCA1 IVS5 + 3A > G. Dis Markers 15(1–3):69–73
Claes K et al (2004) BRCA1 and BRCA2 germline mutation spectrum and frequencies in Belgian breast/ovarian cancer families. Br J Cancer 90(6):1244–1251
King MC, Marks JH, Mandell JB (2003) Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302(5645):643–646
Meijers-Heijboer EJ et al (2000) Presymptomatic DNA testing and prophylactic surgery in families with a BRCA1 or BRCA2 mutation. Lancet 355(9220):2015–2020
Peto J et al (1999) Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 91(11):943–949
Ford M, Murdoch J (1997) The management of ovarian cancer. Br J Hosp Med 58(11):581–583
Couch FJ et al (1996) BRCA2 germline mutations in male breast cancer cases and breast cancer families. Nat Genet 13(1):123–125
Haraldsson K et al (1998) BRCA2 germ-line mutations are frequent in male breast cancer patients without a family history of the disease. Cancer Res 58(7):1367–1371
Johannesdottir G et al (1996) High prevalence of the 999del5 mutation in icelandic breast and ovarian cancer patients. Cancer Res 56(16):3663–3665
Mavraki E et al (1997) Germline BRCA2 mutations in men with breast cancer. Br J Cancer 76(11):1428–1431
Syrjakoski K et al (2004) BRCA2 mutations in 154 Finnish male breast cancer patients. Neoplasia 6(5):541–545
Charef-Hamza S et al (2005) Loss of heterozygosity at the BRCA1 locus in Tunisian women with sporadic breast cancer. Cancer Lett 224(2):185–191
Uhrhammer N et al (2008) BRCA1 mutations in Algerian breast cancer patients: high frequency in young, sporadic cases. Int J Med Sci 5(4):197–202
Ellis D et al (2000) Low prevalence of germline BRCA1 mutations in early onset breast cancer without a family history. J Med Genet 37(10):792–794
Kwon JS et al (2010) Expanding the criteria for BRCA mutation testing in breast cancer survivors. J Clin Oncol 28(27):4214–4220
Ottini L et al (2000) BRCA1 and BRCA2 mutations in central and southern Italian patients. Breast Cancer Res BCR 2(4):307–310
Borg A (2010) Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: the WECARE study. Hum Mut 31(3):E1200-40
Acknowledgments
This project was realized with the funding of an Emmanuel van der Schueren scholarship of the Flemish foundation against cancer to Kim De Leeneer. This research was supported by grant 1.5.150.07 from the Fund for Scientific Research Flanders (FWO) to Kathleen Claes and by GOA grant BOF10/GOA/019 (Ghent University). Bruce Poppe is a senior clinical investigator from FWO.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
De Leeneer, K., Coene, I., Crombez, B. et al. Prevalence of BRCA1/2 mutations in sporadic breast/ovarian cancer patients and identification of a novel de novo BRCA1 mutation in a patient diagnosed with late onset breast and ovarian cancer: implications for genetic testing. Breast Cancer Res Treat 132, 87–95 (2012). https://doi.org/10.1007/s10549-011-1544-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-011-1544-9