
Abstract
Two single nucleotide polymorphisms (SNPs), rs4415084, and rs10941679 on chromosome 5p12 were associated with risk of breast cancer in a recent genome-wide association study (GWAS) of women of European ancestry. Both SNPs are located in a large high-LD region and the causal variant(s) are still unknown. We conducted a nested case–control study in a cohort of African American women to replicate and narrow the region carrying the causal variant(s). We evaluated 14 tagging SNPs in a 98 kb LD block surrounding the index SNPs in 886 breast cancer cases and 1,089 controls from the Black Women’s Health Study. We used the Cochran–Armitage trend test to assess association with breast cancer risk. Odds ratios were derived from logistic regression analyses adjusted for potential confounders including percent European admixture. We confirmed the reported association of rs4415084 SNP with overall risk of breast cancer (P = 0.06), and, as in the original study, observed a stronger association with estrogen receptor positive tumors (P = 0.03). We identified four other SNPs (rs6451770, rs12515012, rs13156930, and rs16901937) associated with risk of breast cancer at the nominal alpha value of 0.05; all of them were located in a 59 kb HapMap YRI LD block. After correction for multiple testing, the association with SNP rs16901937 remained significant (P permutated = 0.038). The G allele was associated with a 21% increased risk of breast cancer overall and with a 32% increase in tumors positive for both estrogen and progesterone receptors. The present results from an African ancestry (AA) population confirm the presence of breast cancer susceptibility genetic variants in the chromosome 5p12 region. We successfully used the shorter range of LD in our AA sample to refine the localization of the putative causal variant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To date, most investigations aimed at identifying common genetic variants associated with breast cancer risk have been carried out in European ancestry (EA) populations. There have been several genome-wide association scans (GWAS) [1–4] and independent replication studies [5–7] of the GWAS findings in EA populations. The variants identified in EA GWAS studies are single nucleotide polymorphisms (SNPs) that tag a large region in a gene (for example intron 2 in the FGFR2 gene [1, 2]) or a gene desert region (for example 8q24 [1]). In this regard, it is important to replicate these findings in other ethnic populations and perhaps use the different linkage disequilibrium (LD) patterns observed in non-European ancestral population to refine associated genomic regions.
As initially demonstrated by Gabriel et al. [8] and further corroborated by the HapMap project (http://hapmap.ncbi.nlm.nih.gov/), populations of African ancestry (AA) have shorter LD blocks on average and more diverse haplotype structure than other ancestral populations. Fine-mapping in an AA population can help to eliminate non-causal variants that cannot be excluded in EA studies due to large regions of high LD in EA populations. We hypothesized that it would be possible to identify common low-penetrance variants associated with breast cancer in AA women and to narrow the regions of interest from findings in EA GWAS by genotyping a dense set of tagging SNPs covering the CEU LD blocks tagged by variants identified in those GWAS. We applied this approach to the region of chromosome 5p12 that was first identified as a region of interest by Stacey et al [9] in their GWAS of an Icelandic population. They found a significant association with breast cancer risk at the genome-wide association level for two SNPs, rs4415084, and rs10941679. Both SNPs are in a 98 kb LD block in HapMap CEU samples and are located more than 100 kb from the nearest gene (MRPS30).
We used DNA samples from participants in the Black Women’s Health Study, an ongoing prospective cohort study of African-American women. We first examined whether the index SNPs from the Icelandic GWAS could be replicated in an AA population. We then examined a dense set of tagSNPs across the CEU LD block containing those index SNPs.
Materials and methods
Study population
We conducted a nested-case control study within the ongoing Black Women’s Health Study (BWHS) [10]. The study began in 1995 when women 21–69 years of age from across the United States completed a 14-page postal health questionnaire. The initial cohort comprised 59,000 women who self-identified as “black” and had a valid address. Follow-up questionnaires are sent every 2 years. Follow-up of the baseline cohort has averaged 80% or greater for each questionnaire.
We used medical records and cancer registry data to confirm self-reported cases of breast cancer, as well as to gain information on tumor characteristics such as estrogen receptor (ER) and progesterone receptor (PR) status. We have obtained records or registry data for 1,151 breast cancer cases reported on the BWHS questionnaires, of which 99.4% were confirmed. Self-reported cases that were disconfirmed have been excluded.
We obtained DNA samples from BWHS participants using the mouthwash-swish method [11]. Approximately 50% of the participants, 27,800 women, provided a sample. Women who provided samples were slightly older than women who did not, but the two groups were similar with regard to educational level, geographic region of residence, body mass index, and family history of breast cancer.
This study includes all cases of breast cancer who provided a DNA sample and were diagnosed through the end of the 2007 follow-up cycle. We selected approximately one matched control per case among BWHS participants who had provided a DNA sample and who were free of breast cancer at the end of the 2007 follow-up period. Controls were matched to cases on year of birth (± 1 year) and geographical region of residence (Northeast, South, Midwest, and West).
The Institutional Review Boards of Boston University and Howard University approved the study protocol.
Selection of tagSNPs and ancestral informative markers
The index SNPs (rs4415084 and rs10941679) are located inside a 98 kb LD block in HapMap CEU samples. We downloaded SNPs covering the entire LD block from the HapMap Yoruba (YRI) database (http://hapmap.ncbi.nlm.nih.gov/). We used the Tagger software [12] implemented in Haploview version 4.1 [13] (http://www.broadinstitute.org/haploview/haploview) to select all tagging SNPs with a minor allele frequency (MAF) ≥ 5% and r 2 ≥ 0.8. The two index SNPS, rs4415084 and rs10941679, were forced into the set. A total of 16 SNPs along the 99 kb LD block were included.
We also selected 30 ancestral informative markers (AIMs) to estimate the percent European ancestry and control for population stratification due to European admixture. The 30 AIMs were selected from a list of validated SNPs in which the top 30 AIMs had allele frequency differences between Africans and Europeans of at least 0.75 [14]. Twenty-nine of the AIMs were successfully genotyped. We used a Bayesian approach as implemented in the Admixmap software [15, 16] to estimate individual admixture proportions. Eighty-one controls included in this breast cancer study had previously been genotyped for a set of 1,536 AIMs as part of an admixture mapping study of a different phenotype. The correlation between percent European admixture determined by our 29 AIMs as compared with the panel of 1,536 AIMs was highly significant (r = 0.87, P < 0.0001), confirming the validity of the smaller set of AIMs.
Genotyping and quality control
The mouthwash-swish saliva samples are stored in -80 degree freezers at the Boston University Molecular Core Genetics Laboratory. DNA was isolated from the samples of breast cancer cases and controls by use of the QIAAMP DNA Mini Kit (Qiagen). Whole genome amplification was performed with the Qiagen RePLI-g Kits using the method of multiple displacement amplification. Amplified samples underwent purification and PicoGreen quantification at the Broad Institute Center for Genotyping and Analysis (Cambridge, MA) before being plated for genotyping.
Genotyping was carried out at the Broad Institute Center for Genotyping and Analysis using the Sequenom MassArray iPLEX technology. Ninety-eight blinded duplicate samples were included to assess reproducibility of the genotypes. An average reproducibility of 99% was obtained among the blinded duplicates. All SNPs with calling rate <90% or a deviation from Hardy–Weinberg equilibrium in the control sample at P < 0.001 were excluded. We also excluded samples with calling rates < 80%. The final analysis included 14 tagging SNPs in 1,975 samples (886 breast cancer cases and 1,089 controls). Mean call rate in the final data set for both SNPs and samples was 99.0%.
Statistical analysis
We tested each SNP for association with breast cancer risk using the Cochran–Armitage trend test of an additive genetic model as implemented in the PLINK software [17]. We used logistic regression analysis (PROC LOGISTIC, SAS statistical software version 9.1.3, SAS Institute Inc., Cary, NC, USA) to estimate per-allele odds ratios, odds ratios for heterozygosity and homozygosity of the high-risk alleles, and 95% confidence intervals. We controlled for age, geographical region of residence (Northeast, South, Midwest, West), birthplace (US, foreign country), and European admixture proportion. To adjust for multiple testing (evaluation of 14 SNPs in the CEU LD block), we used permutation analysis with 100,000 permutations [17]. This method switches the case–control status labels among the individuals to create replicates of the dataset under the null hypothesis. The method generates two sets of empirical P values: an unadjusted value for each individual SNP, and also an adjusted value that takes into account all the SNPs that were tested. Because the permutation approach maintains the LD pattern between the SNPs, it is a better way to control for multiple testing compared to a Bonferroni correction, which assumes independence of the SNPs.
Associations were assessed for all breast cancers together and separately for subtypes of breast cancer defined by ER and PR status. For replication of the EA GWAS finding, we classified cases as either ER positive (+) or ER negative (−) to be consistent with the previously published results. In analyses of other SNPs identified in our genotyping, we also considered PR status. Most cases with hormone receptor status was available were classified as either ER+/PR+ or ER−/PR−. Due to small numbers, results for the other two possible categories, ER+/PR− and ER−/PR+ are not presented.
Results
Table 1 shows characteristics of breast cancer cases and controls. No significant differences were observed in the percentage of European admixture between the groups (19.3% in cases vs. 19.3% in controls).
We observed an association between rs4415084 SNP and risk of breast cancer that is supportive of the result found in the EA GWAS although only marginally significant in our overall sample (Table 2). The association was stronger for cases with ER-positive tumors, with a 25% increase in risk for each copy of the T-allele (p = 0.03). No association was observed for ER-negative tumors. The other previously identified SNP in this region, rs10941679, was not significantly associated with risk of breast cancer overall, or with particular subtypes of tumors defined by ER and PR status. However, the per allele ORs for rs10941679 for breast cancer overall and for ER-positive cancer were similar to the ORs for rs4415084 (Table 2).
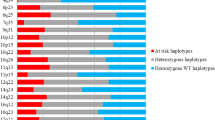
We found four tagging SNPs to be associated with risk of breast cancer at the nominal α = 0.05 level of significance (Fig. 1; Table 3). These tagging SNPs (rs6451770, rs12515012, rs13156930, and rs16901937) are in high LD with each other as measured by D′ (Table 4) and are all located in the second half of the 98 kb CEU LD block (Fig. 1). In the YRI population, that region of the genome appears to be two discrete LD blocks, with all four of the new SNPs residing in the 59 kb block located from 44,714 to 44,773 kb. After adjustment for multiple testing, rs16901937 was the only SNP that remained significant (Table 3). Each copy of the rs16901937 G-allele was associated with a 21% increase in risk of breast cancer. We observed a stronger association with tumors that were positive for both ER and PR receptors; each copy of the rs16901937 G-allele was associated with a 32% increase in risk (Table 3). No significant association was observed with tumors that were negative for both ER and PR receptors.

Scatterplot and LD map of the genotyped tagging SNPs along the 98 kb LD block in the chromosome 5p12 region. The upper panel shows the association results in the logarithmic scale. Positions of the two index SNPs (rs4415084 and rs10941679) are indicated as well as the four newly identified SNPs. The lower panels show the D′ pair-wise values in both CEU and YRI HapMap samples. The four newly identified SNPs are all located in a smaller 59 kb LD block in YRI HapMap samples
Discussion
Our study of AA women from the BWHS confirms the initial findings of the EA GWAS. SNP rs4415084, which was associated with breast cancer risk in the European GWAS conducted by Stacey et al. [9], was associated with breast cancer in BWHS data, overall (P = 0.06) and for ER-positive tumors (P = 0.03). The second SNP, rs10941679, was not statistically significant in BWHS data (P = 0.11 overall and P = 0.10 for ER-positive tumors), but the findings were consistent with a positive association for the same risk allele. These SNPs have been evaluated in two previous, smaller studies of AA women [9, 18]. As a part of replication for the original GWAS, associations were examined in 689 breast cancer cases and 469 controls from a Nigerian case-control study and 428 cases and 457 controls nested in the Multi-ethnic Cohort Study. SNP rs10941679 was not associated with breast cancer in either study, and SNP rs4415084 was associated with breast cancer in the Nigerian study (P = 0.045) but not in the Multi-ethnic Cohort Study. A more recent report [18] evaluated SNP rs10941679 in African Americans and found no association in a combined group of 810 cases and 1,784 controls from two separate studies conducted in the Southern U.S. None of the previous studies of AA populations assessed risk separately according to ER or PR status. Our results add to the evidence that SNP rs4415084 and possibly SNP rs10941679 are tagging a region or regions of importance in the etiology of breast cancer and, in particular, of breast cancers that have estrogen and progesterone receptors.
These two SNPs are located in a 98 kb LD block, stretching from 44,678 to 44,777 kb in the HapMap CEU, which is part of a larger high LD region on chromosome 5p12. The results of our genotyping of additional SNPs have narrowed the region within that CEU block that may contain the true causal variant(s). In particular, we found an association with rs16901937, which resides in the second half of the 98 kb LD block in what is actually a smaller LD block (59 kb) in the YRI population. That smaller block also includes SNP rs10941679 from the original GWAS as well as the other three SNPs (rs6451770, rs12515012, and rs13156930) associated with breast cancer at a nominal level in the BWHS.
The biologic mechanism through which genetic variation in these regions influences breast cancer risk remains unclear. The closest gene is MRPS30, which encodes a component of the mitochondrial ribosome and has been implicated in apoptosis [19, 20]. MRPS30 is also part of a gene expression profile that differentiates ER-positive from ER-negative tumors [21]. As noted above, the associations observed in our study were stronger for ER-positive disease than for ER-negative disease.
A major strength of the current study is the large sample size. With 886 cases and 1,089 controls, this is the largest single study of genetic variation in AA women. Adjustment for multiple comparisons was performed by permutation analysis and the strongest SNP was significantly associated with disease even after adjustment. Cases and controls came from the same base population of AA women who enrolled in the BWHS in 1995. Extensive demographic and risk factor data have been collected from study participants by biennial questionnaires during follow-up. We were able to compare breast cancer cases who provided a saliva sample with those who did not with regard to numerous characteristics and we established that the cases in our analysis were representative of all BWHS cases. In addition, we controlled for potential confounding factors, including European admixture.
The present results from an AA population confirm the importance of the 5p12 region to understanding breast cancer etiology. The findings help to narrow the locus of the true causal regions. Further fine-mapping efforts, whether in AA or other ancestral populations, may be most efficient if focused on these refined genomic regions.
References
Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R et al (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447(7148):1087–1093
Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A et al (2007) A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 39(7):870–874
Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K et al (2009) A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet 41(5):579–584
Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, Platte R, Morrison J, Maranian M, Pooley KA, Luben R et al (2009) Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet 41(5):585–590
Hemminki K, Muller-Myhsok B, Lichtner P, Engel C, Chen B, Burwinkel B, Forsti A, Sutter C, Wappenschmidt B, Hellebrand H et al (2009) Low risk variants FGFR2, TNRC9 and LSP1 in German familial breast cancer patients. Int J Cancer. doi:10.1002/ijc.24986
Antoniou AC, Sinilnikova OM, McGuffog L, Healey S, Nevanlinna H, Heikkinen T, Simard J, Spurdle AB, Beesley J, Chen X et al (2009) Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum Mol Genet 18(22):4442–4456
Garcia-Closas M, Hall P, Nevanlinna H, Pooley K, Morrison J, Richesson DA, Bojesen SE, Nordestgaard BG, Axelsson CK, Arias JI et al (2008) Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet 4(4):e1000054
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M et al (2002) The structure of haplotype blocks in the human genome. Science 296(5576):2225–2229
Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, Jakobsdottir M, Bergthorsson JT, Gudmundsson J, Aben KK et al (2008) Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet 40(6):703–706
Rosenberg L, Adams-Campbell L, Palmer JR (1995) The Black Women’s Health Study: a follow-up study for causes and preventions of illness. J Am Med Womens Assoc 50(2):56–58
Cozier YC, Palmer JR, Rosenberg L (2004) Comparison of methods for collection of DNA samples by mail in the Black Women’s Health Study. Ann Epidemiol 14(2):117–122
de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D (2005) Efficiency and power in genetic association studies. Nat Genet 37(11):1217–1223
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265
Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, Kessing BD, Malasky MJ, Scafe C, Le E et al (2004) A high-density admixture map for disease gene discovery in African Americans. Am J Hum Genet 74(5):1001–1013
McKeigue PM, Carpenter JR, Parra EJ, Shriver MD (2000) Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann Hum Genet 64(Pt 2):171–186
Hoggart CJ, Parra EJ, Shriver MD, Bonilla C, Kittles RA, Clayton DG, McKeigue PM (2003) Control of confounding of genetic associations in stratified populations. Am J Hum Genet 72(6):1492–1504
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
Zheng W, Cai Q, Signorello LB, Long J, Hargreaves MK, Deming SL, Li G, Li C, Cui Y, Blot WJ (2009) Evaluation of 11 breast cancer susceptibility loci in African-American women. Cancer Epidemiol Biomarkers Prev 18(10):2761–2764
Cavdar Koc E, Ranasinghe A, Burkhart W, Blackburn K, Koc H, Moseley A, Spremulli LL (2001) A new face on apoptosis: death-associated protein 3 and PDCD9 are mitochondrial ribosomal proteins. FEBS Lett 492(1–2):166–170
Cavdar Koc E, Burkhart W, Blackburn K, Moseley A, Spremulli LL (2001) The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem 276(22):19363–19374
Van‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT et al (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(6871):530–536
Acknowledgments
We thank the Black Women’s Health Study participants for their continuing participation in this research effort. This work was supported by grants R01CA058420 and R01CA098663 from the National Cancer Institute, Division of Cancer Control and Population Science (http://www.cancercontrol.cancer.gov). The Broad Institute Center for Genotyping and Analysis is supported by grant U54 RR020278 from the National Center for Research Resources (http://www.broadinstitute.org/sections/science/projects/broad/ncrr-center-genotyping-analysis).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ruiz-Narvaez, E.A., Rosenberg, L., Rotimi, C.N. et al. Genetic variants on chromosome 5p12 are associated with risk of breast cancer in African American women: the Black Women’s Health Study. Breast Cancer Res Treat 123, 525–530 (2010). https://doi.org/10.1007/s10549-010-0775-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-010-0775-5