
Abstract
A serious complication associated with breast cancer treatment is the increased risk for development of therapy-related myeloid neoplasms (t-MN). To determine whether dose-intensive adjuvant regimens for breast cancer induce genetic damage to hematopoietic stem cells, defined by the emergence of clonal hematopoiesis, and whether detection of clonal hematopoiesis could be used as an early marker for the subsequent development of t-MN, the Southwest Oncology Group designed a pilot clonality investigation to estimate the incidence of clonal hematopoiesis during and shortly after completion of the dose-intensive neoadjuvant regimens for high-risk breast cancer patients. Peripheral blood samples from 274 patients obtained prior to treatment, at time of surgery, and at 6 and 12 months post-surgery were examined by two different clonality assays: the HUMARA (HUMan Androgen Receptor) assay to estimate the incidence of early genetic damage by clonal proliferation, and microsatellite instability (MSI) testing to screen for LOH or defective DNA mismatch repair mechanisms. Clonal hematopoiesis was negative in 93.5% of the samples analyzed. Five patients showed a HUMARA-positive/MSI-negative pattern, and no patients showed a HUMARA-negative/MSI-positive pattern. With a median follow-up of 3.1 years, one patient in our study developed t-AML at 3 years 5 months after randomization. Our results indicate that clonal hematopoiesis assays performed within the 2 years following dose-intensive neoadjuvant therapy failed to identify an emerging clonal hematopoietic stem cell population. Longer clinical follow-up will be necessary to define better the positive predictive value of detecting clonal hematopoiesis in the HUMARA+/MSI− cases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Therapy-related myeloid neoplasms (t-MN) have been reported with increasing frequency during the last decade in patients receiving intensive neoadjuvant and adjuvant chemotherapy with or without radiotherapy, for treatment of breast cancer [1–7]. Retrospective studies have also suggested that the addition of supportive care measures for these dose-intensive regimens, such as the use of hematopoietic growth factors, may also be associated with an increased risk of t-MN [3, 4, 7]. As disease-free and overall survival for women with advanced-stage breast cancer following treatment with anthracycline–cyclophosphamide–taxane-based regimens and regional local therapy continues to improve, late-effect complications of therapy, including the development of t-MN, must be investigated to maintain and continuously improve efficacy while minimizing life-threatening sequelae [8–10].
Many agents used in the treatment of cancer may induce genetic damage resulting in somatic mutations that confer a selective growth advantage and lead to the expansion of an abnormal clone of hematopoietic stem cells. According to the Jacobs model for leukemogenesis, these acquired somatic mutations are an essential early, possibly initial, step in the development of t-MN, resulting in clonal hematopoiesis with or without any obvious cytogenetic or hematological change, such as dysplasia or cytopenias [11]. The acquisition of additional genetic lesions, such as clonal cytogenetic aberrations, appears to be essential for the subsequent development of t-MN. In support of this model, Carter and others described clonal hematopoiesis in ~ 30% of the 70 clinically asymptomatic patients who had received prior cytotoxic therapy for lymphoma [12, 13]. Using the DNA methylation, X-inactivation-based human androgen receptor assay (HUMARA), Mach-Pascual et al. [14] detected clonality just prior to autologous transplantation in 10 of 78 patients (13.5%) previously treated for non-Hodgkin lymphoma. Four of the 10 patients developed t-MN in comparison to only two of 53 patients with polyclonal hematopoiesis, indicating the HUMARA clonality assay may predict for the development of t-MN (P = 0.004). Moreover, Gale et al. [15] have shown that sequential X-linked clonality assays are predictive of subsequent evolution to frank MDS or AML. These studies suggest that the presence of clonal hematopoiesis following chemotherapy may be a common event and pilot prospective studies are warranted to determine their clinical relevance.
Two common, complementary PCR-based methods to detect clonal hematopoiesis and genetic damage are the HUMARA assay and microsatellite instability (MSI) assay. The highly reproducible HUMARA assay distinguishes polyclonal from monoclonal hematopoiesis based on the highly polymorphic number of tandem CAG repeats (n = 9–36) in the first exon of the HUMARA locus, by differentiating the maternal and paternal X chromosomes and the methylation status of the active and inactive X chromosome [14–18]. The MSI clonality assay screens for the presence of defective DNA mismatch repair mechanisms and loss of heterozygosity (LOH) caused by failure to repair the errors that normally occur during replication of repetitive DNA sequences [19–24]. These alterations are easily observed in the lengths of microsatellites due to deletions or insertions of repeating units to produce novel length alleles in tumor (test) DNA when compared with the normal/germline (control) DNA from the same individual.
With the combined widespread use of adjuvant chemotherapy in breast cancer and the concomitant increase in t-MN, the Southwest Oncology Group designed a pilot prospective investigation (ClinicalTrials.govIdentifier: NCT00016406) to use these clonality assays to estimate the incidence of clonal hematopoiesis during and shortly after completion of the dose-intensive neoadjuvant regimens for high-risk breast cancer patients to address the following questions: (1) Does genetic damage leading to the development of clonal hematopoietic stem cell disorders occur with unacceptable frequency in patients receiving dose-intensive anthracycline-based adjuvant regimens for the treatment of breast cancer? (2) Will careful monitoring of this patient population reveal additional t-MN with long-term follow-up? And, (3) Does the administration of recombinant hematopoietic growth factors, such as G-CSF, used to minimize morbidity and facilitate scheduled drug dosing, play a potentiating role in the development of these secondary malignancies?
Materials and methods
Patients
Peripheral blood samples were obtained from 274 patients registered on a prospective randomized phase III Southwest Oncology Group trial to evaluate neoadjuvant therapy in women with previously untreated inflammatory or locally advanced breast cancer (S0012; ClinicalTrials.govIdentifier: NCT00016406) and who consented for the clonal hematopoiesis correlative study. Briefly, patients were randomized to receive standard doxorubicin (60 mg/m2) and cyclophosphamide (600 mg/m2) given intravenously on day one of a 3-week cycle for five cycles, with paclitaxel (80 mg/m2) given intravenously once a week for 12 weeks (arm 1) versus weekly doxorubicin (24 mg/m2) and daily oral cyclophosphamide (60 mg/m2) for 15 weeks with G-CSF (5 mcg/kg/day) support given on days 2–7 each week followed by weekly paclitaxel (80 mg/m2) for 12 weeks (arm 2) as neoadjuvant therapy for inflammatory and locally advanced breast cancer (Fig. 1). Peripheral blood samples (n = 657) for clonality analyses were collected from February 2002 through September 2007 at the following time points: (a) prior to treatment, (b) at time of surgery, and (c) at 6 and 12 months post-surgery. The clonality assays were performed in a blinded fashion at two separate facilities to obtain the highest degree of confidence.

Protocol Schema for S0012. *After progression and removal from protocol treatment, further therapy is per treating physician’s discretion. **After resection with node dissection, further therapy per treating physician’s discretion
HUMARA assay
Ten to 40 ml of peripheral blood per patient was obtained in sodium heparin tubes. Time in transit for the blood samples ranged from 24 to 96 h. Upon receipt, mononuclear cells were separated by density gradient centrifugation (Ficoll-Paque Plus, Amersham Pharmacia, Piscataway, NJ). T-cells were isolated according to manufacturer’s directions with M 450 Pan-T CD2 Dynabeads (Invitrogen, Carlsbad, CA). T-cell and granulocyte DNA was extracted with standard phenol/chloroform protocols or with the Qiagen EZ-1 BioRobot (Qiagen, Valencia, CA).
The HUMARA assay was performed using a non-radioactive variation of the assay described by Mach-Pascual et al. [14] and quantitated by the method of Delabesse et al. [17]. Briefly, this PCR-based assay amplifies an ~250 to 300 base pair (bp) region of the first exon of the human androgen receptor. Two Hpa II methylation-sensitive sites reside within 100 bp 5′ to the polymorphic CAG repeat. The primers used flanked the methylation-sensitive restriction enzyme sites and the CAG repeat simultaneously. Genomic DNA was digested with RsaI and HpaII enzymes. A known positive control was run with each digestion round for quality control. PCR conditions and primers followed the published protocol; however, a fluorescent (6-FAM) (non-radioactive) labeling method was used. Samples were analyzed on an ABI 3100 with GeneScan software v.3.1 (Applied Biosystems, Foster City, CA). Each reaction was diluted 1:50 in ddH2O. One microliter of this dilution was added to 12 μl Hi Di formamide (Applied Biosystems) with 0.7 μl HD400 ROX size standard (Applied Biosystems) for analysis on the ABI3100. POP-4 polymer was used for all runs. All samples were run in duplicate.
The allelic ratio was defined as the ratio between the two X-linked alleles. Because X-inactivation patterns (XIP) show tissue specificity and only neutrophils, not T-cells, show skewing with age, T-cells collected from each patient served as control tissue to eliminate false positives associated with age-related skewed X-inactivation [25, 26]. Two ratio corrections were used in this study. The corrected ratio, which compensates for preferential amplification that may occur in either T-cell or granulocytic fractions, is the allele ratio of the methylation-sensitive reaction (HpaII and RsaI) divided by the allele ratio of the non-digested (methylation-insensitive) reaction of the same specimen. The second correction, division of the corrected granulocyte ratio by the corrected T-cell ratio, controls for skewed XIP or excessive Lyonization that may mimic true clonal hematopoiesis, especially in older females [27, 28]. Samples were considered skewed if both the T-cell ratio and the granulocyte ratio were above 3, whereas HUMARA-positive samples showed a second corrected ratio (granulocyte/T-cell) greater than 3. All ratios discussed in the results are this final corrected ratio (G/T). Four possible outcomes were observed by the HUMARA assay: negative (polyclonal), positive (monoclonal), monoallelic (non-informative) or skewed XIP. Samples that showed an apparent skewed XIP and a G/T ratio at one time point only were considered indeterminate until a second (follow-up) sample on that patient was processed.
Microsatellite instability testing
Ten MSI markers were chosen for study. The first five MSI markers, BAT26, BAT40, APC, Mfd15CA, and D2S123, were chosen on the basis of the existing literature, documenting their utility in the detection of MSI in a variety of different malignancies. The Big Adenine Tract (BAT)-26 mononucleotide and the D2S123 dinucleotide loci are from the Bethesda reference panel. The other highly informative markers were chosen on the basis of their location in genomic regions where chromosome translocations or LOH have been frequently reported in t-MN and, specifically, where abnormalities have been associated with topoisomerase II inhibitors. These five MSI markers (with genomic location) include AFM240YA11 (3q21), AFM302xb9 (11q23), AFM031xc5 (21q22), AFM337zg5 (12p12), and AFMb298yh5 (20pter-20qter), common “hotspots” in t-MN.
The forward primer of each primer set was 5′-end labeled with a fluorophore to allow for automated fluorescence detection (6Fam, Hex, or Tet: blue, yellow, green fluorescence, respectively). All PCR reactions contained 100 ng of either normal (T-cell) or experimental (PMN) DNA and 2.5 units of AmpliTaq Gold polymerase (Perkin-Elmer, Waltham, MA). “Hot start” PCR amplification was performed according to manufacturer’s recommendations with an extension phase of 45 cycles. MSI analysis was performed using 2 μl of each PCR product and combined with a 350-bp size standard (Perkin-Elmer) labeled with the red fluorescent fluorophore Tamra and run on a 4% polyacrylamide denaturing gel. The gels were run on an ABI 377 instrument and were analyzed following electrophoresis using Genescan Analysis software (Applied Biosystems). Genotyper DNA Fragment Analysis Software (Applied Biosystems) was used to determine the allele sizes of the amplified products.
Statistical analysis
Presence of clonality was compared between the two treatment arms at each time point using Fisher’s exact test. Changes in clonality between successive time points were also explored.
Results
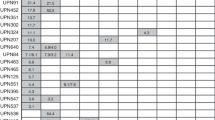
The median age for the 129 patients randomized to arm 1 was 49 years old (range 25–74 years); for patients randomized to arm 2 (145 patients), the median age was 52 years old (range 27–76 years). Follow-up specimens for patients registered to S0012, ranging from one to four per patient (median, 2), were submitted for 198 (72.2%) patients (Fig. 2). Of the 76 patients with only one sample, 51 were removed from protocol therapy for the following reasons: toxicity (n = 14), progressive disease (n = 14), explicit refusal (n = 8) or death prior to completion of sample collection (n = 15). Five patients were removed from protocol therapy because of treatment delays due to infection, EKG abnormalities, or institutional ineligibility. Twenty patients were lost to follow-up.

Number of samples collected consecutively for all patients on study
Overall, the results of both assays were strongly concordant at each time point. In 193/198 patients (316 of 330 or 96% of paired analyses), the MSI and HUMARA assays yielded the same result. None of the samples had evidence of MSI at any of the loci studied at any time point in either arm of the study. In contrast, samples from five of 198 patients had evidence suggesting a clonal hematopoietic population by the HUMARA assay at the following time points: two at pre-treatment, one at 6 months, and two at 12 months. In each of these cases, a polyclonal T-cell fraction and a skewed granulocytic population were noted (Figs. 3, 4). Three of these five patients had follow-up samples showing a polyclonal pre-study pattern with a gradual increase in the corrected ratio over time and the last sample positive for clonal hematopoiesis. One patient was positive at pre-study sample and all follow-up samples. The last patient had a positive pre-study sample but was lost to follow-up. To the best of our knowledge, none of these patients developed microsatellite instability or a hematological malignancy. All remaining patients tested HUMARA-negative or showed polyclonality at each time point tested.

Five patients with HUMARA-positive clonal hematopoiesis. X-axis—sample collection time point, Y-axis—corrected clonal hematopoiesis ratio

Electropherograms from a patient (185528) enrolled on S0012, treatment arm 2. Collection times are indicated at the bottom of the electropherograms. For each collection time, four fractions are analyzed. The top fraction is the T-cell/Rsa I control digestion, the second fraction is the T-cell RsaI/HpaII control digestion, the third fraction is the polymorphonuclear (PMN) fraction/RsaI control digestion, the last fraction is the PMN/RsaI/HpaII test fraction. The two peaks in each fraction represent the two alleles. In the three control fractions, the allele peaks remain relatively constant throughout the collection time points. The test fraction shows a significant decrease in the second allele at both the surgery and 6-month time point
One patient enrolled in the clonal hematopoiesis study developed t-MN at 3 years 5 months after enrollment on the study. This patient was treated on arm 2, and her S0012 pre-treatment sample was polyclonal. By comparing the sequential electropherograms in this patient over 1 year, a gradual shift is observed in her corrected G/T allelic ratios: 1.1 in August 2002, 1.6 in May 2003, and 1.8 in August 2004. Unfortunately, no samples were submitted for this patient in the 20 months prior to diagnosis of t-MN. The sample collected at the time of diagnosis of t-MN (3 years 5 months post-registration) showed a skewed XIP by HUMARA, suggesting clonality; however, a pure T-cell control fraction was not collected at this time point because of her high leukemic blast count, thus precluding the calculation of the second correction factor.
A monoallelic or non-informative HUMARA pattern, defined as a pattern in which the maternal and paternal alleles cannot be distinguished, was observed in 24 (8.6%) patients. This finding is consistent with the ≥10% frequency reported in the literature [14, 27, 29]. Moreover, 43 patients (15.6%) in our study showed non-age-related XIP, a result thought to occur by chance, or other genetic factors involved in the X-inactivation selection process and consistent with the ~20% skewed XIP reported for the general female population [18, 25, 27, 28].
Of the 19 patients (6.9%) who showed HUMARA G/T ratios above 3.0 in one or more samples, 14 patients were classified as “indeterminate” owing to highly skewed XIP in their pre-treatment samples and their corresponding follow-up samples. None of these samples showed MSI. These results were associated with preferential amplification in the granulocyte or the T-cell fraction and consistently observed when the T-cell separation was compromised by the submission of a clotted specimen or the DNA quantity was less than optimal because of low white blood cell counts post-treatment or prolonged specimen transit time.
Discussion
Concerns about the rising incidence of t-MN following dose-intensive neoadjuvant and adjuvant therapy for breast cancer have prompted several large retrospective analyses to identify predisposing risk factors [3, 4, 6, 7, 30, 31]. From the six National Surgical Adjuvant Breast and Bowel Project (NSABP) studies that examined different doxorubicin and cyclophosphamide dosing schedules, t-MN risk correlated more closely with dose intensity than total cumulative dose. Although the overall t-MN risk was small relative to that of breast cancer relapse, dose-intensive doxorubicin (A) and cyclophosphamide (C) regimens that required hematopoietic growth factor support had a cumulative incidence of t-MN of 1.01% at 5 years in comparison to those who received standard AC where the incidence was 0.21%. The NSABP investigators also noted that patients who received breast radiotherapy experienced more t-MN with a cumulative risk of 2.38 (P = 0.006) [7]. A retrospective case–control analysis from France corroborated these findings, noting an increased relative risk of t-MN of 6.3 and 3.9, respectively, for patients receiving G-CSF or radiotherapy [4]. They noted that use of the topoisomerase II inhibitors, particularly mitoxantrone, was associated with the highest risks of t-MN. For patients who received mitoxantrone, the relative risk was 15.6 compared to anthracycline-based regimens. In a separate retrospective analysis, 5510 women over the age of 65 years treated for stages I–III breast cancer who were in the Surveillance, Epidemiology, and End Results (SEER)-Medicare data base were evaluated for the risk of t-MN. The hazard ratio for t-MN for the 906 patients who received hematopoietic growth factors (G-CSF or GM-CSF) during their treatment was 2.14 compared to those who did not receive growth factor support. However, the association of growth factor use with adjuvant chemotherapy and increased incidence of t-MN was not found in another large SEER-Medicare observational analysis [31]. Thus, to address concerns about the relationship of dose-intensive chemotherapy regimens with the use of hematopoietic growth factor support to development of t-MN, we attempted to evaluate some of these identified risk factors in a prospective study. We used two different assays to screen for early evidence of genetic damage to the hematopoietic cell population following two different AC containing regimens (one employing G-CSF support) that were administered as neoadjuvant therapy for women with high-risk breast cancer.
Only one patient in our study cohort has developed t-MN; this patient was treated on arm 2 where patients received continuous exposure to cyclophosphamide and weekly doxorubicin with G-CSF support. Her clonal hematopoiesis test samples at six and 12 months following treatment showed no evidence of an emerging clonal population, and she had normal cytogenetics when she presented with t-MN. Unfortunately, no samples were submitted for this patient in the 20 months prior to her diagnosis of t-MN at 3 years 5 months after randomization. As discussed above, many of the cases of t-MN following chemotherapy for treatment of breast cancer with exposure to toposiomerase II inhibitors typically occur with a short latency period of only 12–24 months following chemotherapy exposure and have been associated with recurring chromosomal translocations involving the MLL gene at chromosome band 11q23. Thus, while we had hypothesized that the median follow-up time would be sufficient to detect the majority of cases of t-MN in this study population, the only case of t-MN occurred later and had a normal karyotype. Thus, our assays were not sufficiently sensitive to detect an early emerging clonal population in this patient. Since the trial design did not include collection of samples for clonality analysis at treatment time points beyond 12 months, it is impossible to know whether these assays may have been able to pick up the emerging population prior to the actual diagnosis of t-MN.
Clonal hematopoiesis, as assessed by serial HUMARA and MSI assays during and following neoadjuvant therapy, was not clearly detected in either arm of S0012. Overall, the two assays were highly concordant. Although there were five cases with evidence of a clonal hematopoietic population using the HUMARA assay, none of these patients had evidence of clonality using the MSI assay. In fact, the HUMARA results in two of these patients suggested that a clonal population may have been present prior to initiation of therapy, whereas two other patients showed a gradually rising ratio suggestive of the emergence of a clonal population during treatment. Importantly, with follow-up now extending 3.1 years, none of these patients have developed t-MN. Plausible alternative explanations for the HUMARA-positive clonality observed in our five patients may be related to the proposals recently reported for the 20–25% of elderly women who show age-dependent skewed XIP by the HUMARA assay, namely, the possible result of either accumulated DNA damage over time or environmentally induced methylation changes suspected to be present in quiescent aging hematopoietic stem cells [32–34]. The relevance of acquired or age-dependent skewed methylation at the HUMARA locus and in our five HUMARA-positive patients remains an intriguing aging hematopoiesis question for future studies, especially in light of a recent study that confirmed the presence of age-related skewing in a sizeable cohort of females aged 43–100 years by three independent X-chromosome inactivation assays [35].
Overall, the clinical results of this study are reassuring regarding the relative safety of this intensive neoadjuvant approach. It is important to note, however, that an additional three cases of t-MN have been reported following S0012 treatment. These patients were enrolled in the clinical trial but did not enroll in our correlative trial to evaluate clonal hematopoiesis. Interestingly, all three of these patients were also enrolled in arm 2 of the study. In one of these patients, t-MN occurred with a short latency period of only 1 year and 5 months following treatment. The other two cases had a similar latency period to the patient in our study cohort, occurring at 3 years 6 months and 3 years 1 month, respectively, following study enrollment. There is no available information about the karyotype of the t-MN in these cases. Thus, a total of four patients randomized to arm 2 of S0012 developed t-MN while, to date, no cases have been reported in patients who received Arm 1 chemotherapy (P = 0.12). Longer clinical follow-up is needed to determine whether the incidence of t-MN will rise because the second group of t-MN associated traditionally with alkylating agents (including cyclophosphamide) peaks from 5 to 7 years following drug exposure.
Owing to the low incidence of t-MN reported in our study cohort enrolled in S0012, our results failed to reveal any characteristic features or mechanisms shown to be predictive for the development of t-MN. Regardless, the findings of the current study suggest that (1) patients with high-risk breast cancers can receive intensive neoadjuvant anthracycline–cyclophosphamide–taxane-based regimens with local radiation with hematopoietic growth factor support without an increased risk of the early development of clonal hematopoiesis or subsequent evolution to t-MN; (2) early detection of clonal hematopoiesis using the methodology described above may not be useful as an early marker of leukemogenesis; or (3) our pre-designated follow-up times were too short to pick up the emergence of the clonal population that would result in t-MN arising with longer latency periods. Analyses of other large prospective studies with long-term follow-up will be needed to provide confirmation or refutation of these possibilities.
References
Vardiman JW, Arber DA, Brunning RD et al (2008) Therapy-related myeloid neoplasm. In: Swerdlow SH, Campo E, Harris NL et al (eds) World Health Organization classification of tumours of haematopoietic and lymphoid tissue. IARC, Lyon
Crump M, Tu D, Shepherd L, Levine M, Bramwell V, Pritchard K (2003) Risk of acute leukemia following epirubicin-based adjuvant chemotherapy: a report from the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1(16):3066–3071. doi:10.1200/JCO.2003.08.137
Hershman D, Neugut AI, Jacobson JS et al (2007) Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst 99(3):196–205. doi:10.1093/jnci/djk028
Le Deley MC, Suzan F, Cutuli B et al (2007) Anthracyclines, mitoxantrone, radiotherapy, and granulocyte colony-stimulating factor: risk factors for leukemia and myelodysplastic syndrome after breast cancer. J Clin Oncol 25(3):292–300. doi:10.1200/JCO.2006.05.9048
Leone G, Pagano L, Ben-Yehuda D, Voso MT (2007) Therapy-related leukemia and myelodysplasia: susceptibility and incidence. Haematologica 92(10):1389–1398. doi:10.3324/haematol.11034
Praga C, Bergh J, Bliss J et al (2005) Risk of acute myeloid leukemia and myelodysplastic syndrome in trials of adjuvant epirubicin for early breast cancer: correlation with doses of epirubicin and cyclophosphamide. J Clin Oncol 23(18):4179–4191. doi:10.1200/JCO.2005.05.029
Smith RE, Bryant J, DeCillis A, Anderson S (2003) Acute myeloid leukemia and myelodysplastic syndrome after doxorubicin-cyclophosphamide adjuvant therapy for operable breast cancer: the National Surgical Adjuvant Breast and Bowel Project Experience. J Clin Oncol 21(7):1195–1204. doi:10.1200/JCO.2003.114
Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365(9472):1687–1717. doi:10.1016/S0140.6736(05)66544-0
Mariotto AB, Feuer EJ, Harlan LC, Abrams J (2006) Dissemination of adjuvant multiagent chemotherapy and tamoxifen for breast cancer in the United States using estrogen receptor information: 1975–1999. J Natl Cancer Inst Monogr (36):7–15. doi:10.1093/jncimonographs/lgj0003
Trudeau M, Charbonneau F, Gelmon K et al (2005) Selection of adjuvant chemotherapy for treatment of node-positive breast cancer. Lancet Oncol 6(11):886–898. doi:10.1016/S1470-2045(05)70424-1
Jacobs A (1991) Leukaemia Research Fund annual guest lecture 1990. Genetics lesions in preleukaemia. Leukemia 5(4):277–282
Abrahamson G, Fraser NJ, Boyd J, Craig I, Wainscoat JS (1990) A highly informative X-chromosome probe, M27 beta, can be used for the determination of tumour clonality. Br J Haematol 74(3):371–372. doi:10.1111/j.1365-2141.1990.tb02601.x
Carter G, Hughes DC, Clark RE et al (1990) RAS mutations in patients following cytotoxic therapy for lymphoma. Oncogene 5(3):411–416
Mach-Pascual S, Legare RD, Lu D et al (1998) Predictive value of clonality assays in patients with non-Hodgkin’s lymphoma undergoing autologous bone marrow transplant: a single institution study. Blood 91(12):4496–4503. doi:0006-4971/98/9112-0011$3.00/0
Gale RE, Bunch C, Moir DJ, Patterson KG, Goldstone AH, Linch DC (1996) Demonstration of developing myelodysplasia/acute myeloid leukaemia in haematologically normal patients after high-dose chemotherapy and autologous bone marrow transplantation using X-chromosome inactivation patterns. Br J Haematol 93(1):53–58. doi:10.1046/j.1365-2141.1996.4751014.x
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51(6):1229–1239
Delabesse E, Aral S, Kamoun P, Varet B, Turhan AG (1995) Quantitative non-radioactive clonality analysis of human leukemic cells and progenitors using the human androgen receptor (AR) gene. Leukemia 9(9):1578–1582
Gale RE, Mein CA, Linch DC (1996) Quantification of X-chromosome inactivation patterns in haematological samples using the DNA PCR-based HUMARA assay. Leukemia 10(2):362–367
Bocker T, Diermann J, Friedl W et al (1997) Microsatellite instability analysis: a multicenter study for reliability and quality control. Cancer Res 57(21):4739–4743
Canzian F, Salovaara R, Hemminki A et al (1996) Semiautomated assessment of loss of heterozygosity and replication error in tumors. Cancer Res 56(14):3331–3337
Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R (1992) Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics 12(2):241–253. doi:0888-7543/923.00
Fey MF (1997) Microsatellite markers in leukaemia and lymphoma: comments on a timely topic. Leuk Lymphoma 28(1–2):11–22. doi:10.3109/10428199709058325
Hoang JM, Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R (1997) BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res 57(2):300–303
Zhou XP, Hoang JM, Cottu P, Thomas G, Hamelin R (1997) Allelic profiles of mononucleotide repeat microsatellites in control individuals and in colorectal tumors with and without replication errors. Oncogene 15(14):1713–1718
Gale RE, Wheadon H, Boulos P, Linch DC (1994) Tissue specificity of X-chromosome inactivation patterns. Blood 83(10):2899–2905
Chen GL, Prchal JT (2007) X-linked clonality testing: interpretation and limitations. Blood 110(5):1411–1419. doi:10.1182/blood-2006-09-018655
Busque L, Mio R, Mattioli J et al (1996) Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood 88(1):59–65
Tonon L, Bergamaschi G, Dellavecchia C et al (1998) Unbalanced X-chromosome inactivation in haemopoietic cells from normal women. Br J Haematol 102(4):996–1003. doi:10.1046/j.1365-2141.1998.00863.x
Boudewijns M, van Dongen JJ, Langerak AW (2007) The human androgen receptor X-chromosome inactivation assay for clonality diagnostics of natural killer cell proliferations. J Mol Diagn 9(3):337–344. doi:10.2353/jmoldx.2007.060155
Smith RE (2003) Risk for the development of treatment-related acute myelocytic leukemia and myelodysplastic syndrome among patients with breast cancer: review of the literature and the National Surgical Adjuvant Breast and Bowel Project experience. Clin Breast Cancer 4(4):273–279
Patt DA, Duan Z, Fang S, Hortobagyi GN, Giordano SH (2007) Acute myeloid leukemia after adjuvant breast cancer therapy in older women: understanding risk. J Clin Oncol 25(25):3871–3876. doi:10.1200/JCO.2007.12.0832
Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL (2007) Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447(7145):725–729. doi:10.1038/nature05862
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA (2007) Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol 5(8):e201. doi:10.1371/journal.pbio.0050201
Choi JK, Kim SC (2007) Environmental effects on gene expression phenotype have regional biases in the human genome. Genetics 175(4):1607–1613. doi:10.1534/genetics.106.069047
Busque L, Paquette Y, Provost S, Roy DC, Levine RL, Mollica L, Gilliland DG (2009) Skewing of X-inactivation ratios in blood cells of aging women is confirmed by independent methodologies. Blood 113(15):3472–3474. doi:10.1182/blood-2008-12-195677
Acknowledgments
This investigation was supported in part by a grant from the Department of Defense, DOD # DAMD17-97-1-7088 and in part by the following PHS Cooperative Agreement grant numbers awarded by the National Cancer Institute, DHHS: CA32102, CA38926, CA58882, CA86780, CA14028, CA20319, CA52654, CA63845, CA379891, CA63844, CA22433, CA35431, CA76448, CA35176, CA58861, CA46441, CA35178, CA67575, CA45808, CA12644, CA11083, CA35192, CA76447, CA46282, CA45560, CA04919, CA45377, CA35090, CA74811, CA95860, CA35281, CA74647, CA76462, CA35128, CA76462, CA46136, and CA35119. The authors also thank Mr. Aaron Theisen for assistance in paper preparation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Slovak, M.L., Bedell, V., Lew, D. et al. Screening for clonal hematopoiesis as a predictive marker for development of therapy-related myeloid neoplasia (t-MN) following neoadjuvant therapy for breast cancer: a Southwest Oncology Group study (S0012). Breast Cancer Res Treat 119, 391–398 (2010). https://doi.org/10.1007/s10549-009-0597-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-009-0597-5