
Abstract
Meta-analyses of microarray data indicate that GATA3 is co-expressed with estrogen receptor alpha (ER) in breast cancer cells. While the significance of this remains unclear, it is thought that GATA3 may serve as a prognostic indicator in breast tumors and may play a role in ER signaling. Recently, reciprocal regulation of GATA3 and ER transcription was demonstrated, suggesting that control of their expression is intertwined. We sought to determine whether GATA3 and ER expression was also coordinately regulated at other levels. Unlike ER, GATA3 was not under epigenetic control and was not re-expressed in the presence of DNMT or HDAC inhibitors in ER/GATA3-negative cells. However, like ER, these inhibitors decreased GATA3 expression in ER/GATA3-positive cell lines. We have previously reported that ER mRNA stability is increased through binding of the RNA-binding protein HuR/ELAV1 to the 3′untranslated region (UTR) and that DNMT and HDAC inhibitors reduce ER expression by altering this interaction. Biotin pull-down assays using a biotinylated GATA3 RNA probe confirmed that HuR also binds to the GATA3 3′UTR. Inhibition of HuR using siRNA probes decreased GATA3 mRNA, mRNA stability and protein expression, indicating that HuR plays a role in regulating GATA3 expression. Inhibition of either HuR or GATA3 reduced cell growth of MCF7 cells. Based on our findings, it is clear that coordinate regulation of ER and GATA3 occurs, however differences do exist. These findings may aid in identification of new targets that control cell growth of breast cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microarray analyses of primary invasive breast carcinomas have shown the coordinate expression of GATA3 along with estrogen receptor alpha (ER) and suggested a role for GATA3 as a prognostic indicator in breast cancer [1–7]. Although this coordinate expression and the importance of GATA3 in mammary cell differentiation have been well documented, the role of GATA3 in breast cancer is less clear. GATA3 is necessary for luminal cell differentiation, and lack of GATA3 expression may result in breast cancer progression and metastasis [8–10]. The recent discovery of the coordinate expression of GATA3 along with ER in primary breast cancers has led to speculation that GATA3 may be involved in the control of ER expression. At the molecular level, the nature of the interaction between GATA3 and ER is becoming clearer. Eeckhouse et al. [11] recently demonstrated a role for GATA3 in the transcriptional activation of ER in the T47D breast cancer cell line mediated through binding of GATA3 to an enhancer region upstream of ER. The need for a more complete understanding of the role of GATA3 in the regulation of ER and in breast cancer, however, is still warranted.
Since ER and GATA3 appear to be coordinately expressed, in this study, we sought to determine whether GATA3 was regulated at the molecular level similarly to that of ER in breast cancer cells. Regulation of ER is complex and occurs at the epigenetic, transcriptional and post-transcriptional levels [12–17]. In breast cancer cells, lack of ER expression can result from epigenetic silencing of the gene. Changes in DNA methylation of the CpG island within the promoter and first exon, along with changes in histone modifications including histone acetylation, result in ER silencing [18]. These processes are reversible; treatment with pharmacologic agents that inhibit the activity of DNA methyltransferase, such as 5-aza 2′ deoxycytidine (AZA), or histone deacetylase, such as trichostatin A (TSA), re-establish expression of ER mRNA and produce functional ER protein in ER-negative cells [19]. Re-expression of functional ER can dramatically impact therapeutic options, including treatment with tamoxifen. Tamoxifen treatment inhibits cell proliferation by preventing activation of ER protein [19]. Indeed, in ER-negative breast cancer cell lines that are de novo resistant to tamoxifen due to the lack of ER expression, treatment with such agents restores sensitivity to tamoxifen. This occurs in ER− cells through increased transcriptional activation of ER. AZA and/or TSA treatment alters the histone code surrounding the ER gene, forming the same transcription factor complexes that bind to the promoter and induce transcriptional activation of ER in ER-positive cell lines [20–22]. This suggests a close link between epigenetic and transcriptional control of ER expression in breast cancer cell lines.
Control of ER expression also occurs at the post-transcriptional level. We along with others have shown that the 3′untranslated region (3′UTR) of ER contains elements that modulate ER mRNA levels. Altering the association of RNA-binding proteins, including the RNA-binding protein HuR, and miRNAs can impact ER mRNA stability and protein levels [15–17, 23–26]. Our previous work has demonstrated that HuR binds to the ER 3′UTR and increases its mRNA stability. This is consistent with HuR’s role in increasing mRNA stability of transcripts to which it is bound. We have also shown that treatment with AZA and TSA results in the opposite effect on overall ER expression in ER+ compared with ER− cells. Whereas in ER—(i.e. MDA-MB-231 cells) AZA/TSA treatment results in epigenetic re-expression of ER, treatment of ER + MCF7 cells with the same agents reduced ER mRNA and protein levels by decreasing the association of the RNA-binding protein, HuR with the ER 3′UTR [17]. While these drugs are known to activate gene expression, we have shown that they also alter the subcellular localization of HuR and in doing so decreases ER mRNA stability [16, 17]. Taken together, these data suggest that ER expression is tightly regulated and involves control at many molecular levels. Given that GATA3 and ER are coordinately expressed, this provides potential mechanisms that may also control GATA3 expression as well.
Since expression of ER and GATA3 appears to be coordinated in breast cancer, we sought to determine whether GATA3 expression was regulated similarly as that of ER (epigenetic, post-transcriptional) in breast cancer cell lines to further establish whether GATA3 could contribute to the molecular control of ER expression. Our data is the first to demonstrate that HuR stabilizes GATA3 mRNA, and that expression of GATA3, like ER, is complex and involves regulation at many levels.
Materials and methods
Cell culture, maintenance and treatment
MCF7, MDA-MB-231 and BT474 cells were obtained from the ATTC (Manassas, VA) cultured according to the suppliers protocol at 37°C, 5% CO2 in DMEM (Invitrogen, Carlsbad, CA) media supplemented with 5% fetal bovine serum (Invitrogen, Carlsbad, CA), 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA) and 1% GlutaMax (Invitrogen, Carlsbad, CA). Unless otherwise stated, cells were plated at 500,000 cells/10 cm plate. Treatments included AZA (2.5 μM; Sigma, St. Louis, MO) or TSA (100 ng/ml; Sigma, St. Louis, MO).
Transient transfection
Cells were transfected with siRNA probes specifically targeting HuR expression as previously described [16, 17]. Nonsense siRNA probes were used to control for non-specific interactions. All probes were purchased from Applied Biosystems and transfected into MCF7 or BT474 cells according to manufacturer’s protocol. Briefly, siRNA probes were diluted in OPTI-MEM (Invitrogen, Carlsbad, CA) media, mixed with siPORT (Ambion, Austin, TX) transfection reagent and allowed to incubate at room temperature for 15–20 min. siPORT/siRNA mixtures were added to cells plated in 24-well plates. Cells were placed in the incubator in low serum OPTI-MEM media for 4 h prior to adjusting serum concentrations to normal levels to allow for transfection of siRNA probes to occur. After 72–96 h of transfection, inhibition of gene expression was confirmed by real-time RT–PCR and Western blotting. Rescue experiments were performed by silencing HuR via transfection of siRNA probes on day 1, followed by transfection of wild type HuR plasmids on day 2. RNA was then harvested on day 4. Wild type HuR plasmids, kindly provided by Dr. Myriam Gorospe, have been successfully used previously in this cell line [16].
RNA electrophoretic mobility shift assay
RNA electrophoretic mobility shift assay (REMSA) analysis was performed using cytoplasmic extracts from MCF7 cells as previously described [16]. To each binding assay, equal amounts of extract, 2–6 μg, RNA-binding buffer (10 mM Tris, pH 7.5, 1.5 mM MgCl2, 250 mM KCl, 0.5 mM DTT, 2 μg/ml leupeptin, 0.5% aprotinin) and 0.5 ng biotinylated probe was added and left to incubate at room temperature for 20 min. Heparin was added for an additional 10 min to minimize non-specific protein binding. Complexes were then separated on 5% non-denaturing acrylamide gels in 0.5% TBE buffer at 100 volts for up to 1–2 h and transferred to nylon membrane at 100 v for 30 min. RNA–protein complexes were stabilized by UV crosslinking the membrane at 254 nm for 10 min and probed with stabilized streptavidin–HRP conjugate. Complexes were visualized by chemiluminescence (Pierce Chemiluminescent Nucleic Acid Detection Kit, Pierce, Rockford, IL). Supershift assays included incubation of protein–probe reactions with excess HuR primary antibody (2 μg) or a non-specific control antibody, GAPDH, for 10 min at room temperature prior to separation on the gel.
Probe synthesis and biotin pull-down assays
Probes for biotin pull-down analysis were synthesized using PCR primers specific for the GATA and ER 3′UTR that contain a 5′ T7 tag to facilitate biotinylation as previously described [16]. The following PCR primers were used: GATA3 UTR sense: T7- TCA CAG GGC CCC CAG C; UTR antisense: CGG CAA CTG GTG AAC GGT AAC; ER UTR1 sense: T7- ATT CCT ATG GCA ATG CAT CCT TTT A; UTR1 antisense: CCC AGG GCT AAA TGC AAC A; UTR2b sense: T7- CTA CTC AGG CTG ACT GGG G; UTR2 antisense: GAA AGT AGG GCA GAA ACT GGA TA. The ER UTR1 probe was used previously and has shown HuR binding. In this assay, ER UTR1 was used as a positive control. ER UTR2b is a region that does not bind HuR and is used as a negative control in this assay. Probes were generated by PCR at an annealing temperature of 64°C for 1.5 min with a 2 min extension at 72°C. Biotinylated probes were synthesized using the MAXIscript T7 kit (Ambion, Austin, TX) with Biotinylated dCTP (Enzo Life Sciences, Plymouth Meeting, PA) incorporated. For pull-down assays, cytoplasmic proteins were incubated with biotinylated probe at room temperature for 30 min with slow mixing. Streptavidin beads (Invitrogen, Carlsbad, CA) were then added for 30 min at room temperature. Beads were then washed extensively in cold PBS prior to addition of Laemmli’s buffer and Western blotting. Probe only (no lysates added) was also examined to exclude any non-specific binding.
Western blotting
Whole cell lysates, cytoplasmic or nuclear extracts were harvested as previously described using a modification of the Schreiber method [27]. Briefly, following treatment cells were washed in cold PBS. Cellular membranes were lysed in 400 μl Buffer A (10 mM Hepes, 1 mM DTT, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM KCl, 0.1 mM PMSF, 1 μl protease inhibitors) prior to addition of 0.625% NP-40. Cells were spun for 30 s, and cytoplasmic extracts were removed from the remaining nuclear pellet. Nuclear pellets were re-suspended in Buffer C (20 mM Hepes, 1 mM DTT, 1 mM EDTA, 1 mM EGTA, 420 mM KCl and 1 μl protease inhibitors) and frozen at −80°C until use. A total of 40 μg cytoplasmic protein or 5 μg nuclear protein were separated on 12% PAGE gels (NuSep Gels, ISC Bioexpress, Kaysville, UT) and transferred to nitrocellulose membranes (BioRad, Hercules, CA) for analysis. Membranes were hybridized with primary antibody (HuR3A2 sc5261, Santa Cruz, Santa Cruz, CA, 1:2000; GATA3, Cell Signaling, Danvers, MA, 1:2000; calnexin SPA-865F, StressGen, Ann Arbor, MI; Actin, A4700 Sigma, St. Louis, MO 1:10,000) overnight at 4°C and developed using the enhanced chemilumiscence (ECL) Plus system (GE Healthcare, NY, NY). Band intensity was determined using densitometry and Image Quant software (GE Healthcare, NY, NY).
Reverse transcription and real-time PCR
RNA was harvested from cells using TriZOL reagent (Invitrogen, Carlsbad, CA) as previously described [28]. cDNA was synthesized from 3 μg total RNA using Superscript reverse transcriptase enzyme (Invitrogen, Carlsbad, CA) at 37°C for 1 h. Real-time PCR was performed on the 7900 Fast RealTime PCR machine (Applied Biosystems, Carlsbad, CA) and TaqMan Assay kits (Applied Biosystems, Carlsbad, CA) that include primers and FAM-labeled probe sets specifically targeting ER, GATA3, HuR and GAPDH. Real-time PCR was conducted using standardized conditions with a 60°C annealing temperature for 30 s. All primers used were validated real-time PCR primers purchased from Applied Biosystems (Carlsbad, CA).
Results
GATA3 expression correlates with ER expression in breast cancer cell lines
Several studies have shown that GATA3 expression correlates with ER expression in primary breast tumors. We confirmed this association in well-characterized breast cancer cell lines. GATA3 mRNA and protein was detected in ER-positive MCF7 and BT474 cells, but not found in ER-negative MDA-MB-231 or Hs578t cells (Fig. 1).

GATA3 expression is correlated with ER expression in breast cancer cell lines. GATA3 mRNA and protein levels were evaluated in MDA-MB-231, Hs578t, MCF7 and BT474 cells. Both mRNA and protein expression were correlated with ER expression in these cell lines. Shown are a real-time PCR (average of 3 experiments normalized to GAPDH expression) and b representative Western Blot. Actin was used as a loading control
GATA3 is not epigenetically regulated
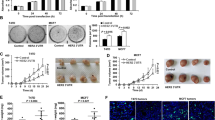
It is well established that ER is epigenetically regulated in breast cells. Epigenetic silencing occurs when cytosine methylation and histone deacetylation of the ER CpG island and promoter region is detected. Treatment of cells with pharmacologic agents that reverse these modifications, including AZA and TSA, also re-establish transcriptional activation of the underlying gene. Existence of a CpG island and transcriptional re-expression of ER through inhibition of these epigenetic processes are evident for ER in human breast cancer [18, 29–32]. Therefore, we searched for evidence of a CpG island (defined as greater than 100 bp, greater than 50% GC and greater than 60% observed/expected ratio) in the promoter region of GATA3 using the MethPrimer program [33]. Evidence of a CpG island would suggest potential epigenetic regulation of GATA3. Interrogation of the GATA3 promoter region revealed two putative CpG islands that are greater than 200 bp in length and greater than 60% GC rich (Fig. 2a marked in the colored region). We next tested whether inhibition of DNA methylation and histone deacetylation could restore expression of GATA3 mRNA as inhibition of these processes restores ER expression [17]. MDA-MB-231, MCF7 and BT474 cells were treated with the DNA methyltransferase inhibitor 5-aza 2′ deoxycytidine (2.5 μM, AZA) and histone deacetylase inhibitor trichostatin A (100 ng/ml, TSA). These agents administered at these doses, either alone or in combination, have been shown to restore expression of ER in ER-negative cell lines [18] (Fig. 2b). In ER/GATA3-negative MDA-MB-231 cells, AZA/TSA treatment had no effect on GATA3 mRNA levels, whereas in ER/GATA3-positive MCF7 and BT474 cells, AZA/TSA treatment significantly decreased GATA3 mRNA and protein expression (Fig. 2b–e). This decrease in GATA3 expression is consistent with our previous findings in ER+ cells [17]. AZA/TSA treatment of MCF7 cells reduced ER mRNA, mRNA stability and protein expression. This decrease resulted from decreased cytoplasmic levels of the RNA-binding protein HuR that binds to the ER 3′UTR and increases its mRNA stability. Indeed, evaluation of the GATA3 gene sequence revealed that the GATA3 3′UTR is extremely AU rich and encodes nine canonical HuR-binding sites, suggesting that GATA3 mRNA expression could be regulated at the post-transcriptional level through interactions with RNA-binding proteins (Fig. 6). Therefore, we focused our attention on investigating control of GATA3 mRNA stability and the potential involvement of HuR.

GATA3 is not epigenetically regulated, but AZA and TSA treatment decreases expression in GATA3+ cells. a Evidence of a CpG island was evaluated using MethPrimer software [33] GATA3 has 2 large CpG islands within the promoter region, marked in the colored region (from 100bp–300bp and 400bp–725bp as shown on this graph), including one surrounding the transcriptional start site (tss, marked with the arrow). The CpG island was defined as having greater than 50% CG content and greater that 0.6 observed/expected ratio. b AZA and TSA treatment of MDA-MB-231 cells did not result in epigenetic re-expression and increase GATA3 mRNA expression (top panel). Treatment of the same cell line with AZA/TSA did result in the epigenetic re-expression of ER (bottom panel). Shown is the average of 3 real-time RT–PCR assays using GAPDH as normalization controls. c AZA/TSA treatment of GATA3 + MCF7 cells reduced GATA3 mRNA levels. This is a similar finding to ER expression in MCF7 cells following AZA/TSA treatment. d AZA/TSA treatment of GATA3 + BT474 cells reduced GATA3 mRNA levels. e Reduction in GATA3 mRNA correlated with a decrease in GATA3 protein. MCF7 and BT474 cells were treated with AZA/TSA, and cytoplasmic extracts were harvested. GATA3 expression decreased in both MCF7 and BT474 cells as demonstrated by Western blotting. GPADH was used as a loading control
HuR increases GATA3 mRNA stability
To determine whether HuR contributed to GATA3 mRNA stability, we treated MCF7 and BT474 cells with siRNA probes specifically silencing HuR expression. Silencing of HuR decreased GATA3 mRNA levels in both cell lines (Fig. 3a, b). Among its many functions, HuR can bind to the 3′untranslated region (UTR) and increase mRNA stability [34]. To test whether inhibition of HuR altered GATA3 mRNA stability, MCF7 cells were treated with siRNA probes specifically silencing HuR while nascent transcription was inhibited with actinomycin D. Inhibition of HuR significantly decreased GATA3 mRNA stability (t ½ = 1.5 h nonsense vs 0.5 h siHuR)(Fig. 3c). Overexpression of wild type HuR in siHuR transfected MCF7 cells was able to rescue GATA3 expression, restoring mRNA levels close to control levels (Fig. 3d).

The RNA-binding protein HuR increases GATA3 mRNA expression and stability. a Silencing of HuR using siRNA probes decreased GATA3 mRNA levels in both MCF7 and BT474 cells (top panel) and protein level (bottom panel). Degree of HuR silencing obtained is shown by Western blotting. b Silencing of HuR decreased GATA3 mRNA stability (P < 0.01). After silencing of HuR using siRNA probes in MCF7 cells, nascent RNA transcription was stopped using Actinomycin D. RNA was harvested every hour and amount of GATA3 mRNA assessed by real-time RT–PCR. c Overexpression of HuR in the presence of siRNA probes silencing HuR rescues GATA3 expression in MCF7 cells. Cells were transfected with siRNA probes or siRNA probes + wild type HuR plasmids. GATA3 expression was determined by real-time PCR. GAPDH was used as a loading control
HuR binds to the GATA3 3′UTR
We next tested whether HuR binds to the GATA3 3′UTR. Using biotin pull-down assays, we found that HuR binds to the GATA3 3′UTR region in MCF7 cells (Fig. 4a). This interaction was eliminated with the inhibition of HuR using siRNA probes (Fig. 4b). REMSA analysis using the same UTR probe further confirmed the interaction of HuR and GATA3 3′UTR as the complex that formed along the GATA3 3′UTR was shifted with addition of the HuR antibody but not with a non-specific antibody, GAPDH (Fig. 4c).

HuR binds to the GATA3 3′UTR. a Biotin pull-down assay using GATA3 3′UTR probes followed by Western blotting for HuR. Co-incubation of cytoplasmic extracts with biotinylated GATA3 3′UTR probes shows that HuR binds readily to the 3′UTR. Lane 1 is a western control of cytoplasmic extracts, lane 2 is a bead-only control, lane 3 is a lysate only control and lane 4 is cytoplasmic extract + probe. b Inhibition of HuR in MCF7 cells eliminates interaction with the GATA3 3′UTR probe. Cytoplasmic extracts from siHuR or nonsense transfected MCF7 cells were incubated with the GATA3 biotinylated 3′UTR probe. HuR interacts with the GATA3 3′UTR in nonsense control extracts but is not present in siHuR treated CE. Lane 1 is a western control of cytoplasmic extracts, lane 2 contains CE from nonsense transfected MCF7 cells and lane 3 contains CE from siRNA transfected MCF7 cells silencing HuR expression. ER probes were used as a negative (lane 1) and positive (lane 2) control for HuR binding in CE from MCF7 cells. HuR binds to the UTR1b region, but not to the UTR2b region. c RNA electrophoresis mobility assay (REMSA) using the biotinylated GATA3 3′UTR probe and cytoplasmic extracts from MCF7 cells. A complex was formed along the GATA3 3′UTR. This complex shifted in the presence of the HuR antibody, but not when supershifted using a non-specific GAPDH antibody
Inhibition of HuR or GATA3 decreases cell proliferation in MCF7 cells
Inhibition of HuR or GATA3 for 72 h reduced cell counts by 35 and 44%, respectively, indicating that HuR and GATA3 are necessary for cell proliferation. Inhibition of HuR and GATA together did not further reduce cell counts (Fig. 5).

Inhibition of HuR or GATA3 decreases MCF7 cell growth. Inhibition of HuR or GATA3 using siRNA probes silencing HuR or GATA3 significantly reduced cell counts in MCF7 cells. Inhibition of both together did not further decrease cell growth. Shown is the average of 3 experiments done in triplicate
Discussion
Like ER, regulation of GATA3 expression is complex and occurs at many levels. While ER and GATA3 can be detected coordinately and their expression is clearly linked in primary and breast cancer cell lines, clear mechanistic differences in the regulation of these genes exist.
Epigenetic regulation of ER is one key mechanism that controls the overall expression of ER in breast cancer cells. Treatment of ER-negative cancer cells with DNA methyltransferase and histone deacetylase inhibitors results in the epigenetic re-expression of ER mRNA and re-establishment of functional ER protein. These agents alter the cytosine methylation and histone acetylation patterns within the CpG island located in the promoter and first exon of the ER gene. Similar to the ER promoter, the GATA3 promoter also encodes a large CpG island suggesting it too may be regulated at the epigenetic level (Fig. 2a). Interestingly, when we treated with the same DNA methyltransferase and histone deacetylase inhibitors that result in ER re-expression in ER-negative MDA-MB-231 cells, 5-aza- 2′ deoxycytidine (AZA) and trichostatin A (TSA), respectively, we did not detect any change in GATA3 mRNA levels (Fig. 2b). Clearly, at this dosage and timeframe, ER expression is re-established (Fig. 2b). Therefore, while ER is under epigenetic control in breast cancer cells, GATA3 expression is not. Further, re-establishment of ER expression and function using these epigenetic mediators does not appear to require the coordinate re-expression of GATA3.
We previously reported that treatment of ER-positive breast cancer cell lines with AZA and TSA reduced ER mRNA and protein expression in these cells [16, 17]. In these breast cancer cell lines, AZA/TSA treatment decreased the cytoplasmic levels of HuR, decreased the association of HuR with the ER 3′UTR and reduced ER mRNA stability [16, 17]. We hypothesized that like ER, GATA3 mRNA could be stabilized by the binding of the RNA-binding protein HuR with the GATA3 3′UTR. This was based on a several findings. Like ER, (1) GATA3 mRNA and protein expression was decreased following AZA/TSA treatment of GATA3-positive breast cancer cells (Fig. 2c, d). (2) The 3′UTR of GATA3 is AU rich, suggesting it could be regulated through interactions with RNA-binding proteins (Fig. 6), and (3) The GATA3 3′UTR encodes 9 canonical HuR-binding sites, NNUUNNUUU (Fig. 6). Indeed, we found that HuR does play a significant role in stabilizing GATA3 mRNA (Fig. 3). Inhibition of HuR significantly reduced GATA3 mRNA expression in both MCF7 and BT474 cell lines (Fig. 3a) through decreasing GATA3 mRNA stability (Fig. 3b) and decreasing the interaction of HuR with the GATA3 3′UTR (Fig. 4).

The GATA3 3′UTR contains several HuR consensus sequences. HuR has been shown to bind to the consensus sequence NNUUNNUUU. Evaluation of the GATA3 3′UTR sequences shows that 9 HuR consensus sequences are encoded within this region. Putative HuR-binding sites are in bold and underlined
The finding that ER and GATA3 are both regulated at the RNA level by the RNA-binding protein, HuR, led us to test whether inhibition of HuR, GATA3 or both together would reduce cell proliferation in ER/GATA3+ cell lines. We found that there was a 35% decrease in cell proliferation in the absence of HuR expression in MCF7 cells. This was similar to the decrease in proliferation found in the absence of GATA3 (44%). Inhibiting both GATA3 and HuR together, however, did not lower proliferation rates (34% reduction). Since a further decrease in proliferation following silencing of both GATA3 and HuR was not detected, we feel that HuR may be central to controlling GATA3-mediated cell growth and that inhibition of HuR is sufficient to inhibit GATA3 controlled proliferation.
Clearly, overlapping mechanisms controlling expression of GATA3 and ER exist. However, these mechanisms are not completely redundant, suggesting that independent regulation of either gene could occur. Since GATA3 re-expression is not required for ER expression, as seen by lack of epigenetic re-expression, GATA3 regulation of ER mRNA may not be essential to restore ER functionality. However, since AZA/TSA-mediated ER re-expression is readily detectable at the RNA level and less so at the protein level, we cannot rule out that GATA3 may enhance ER mRNA and/or protein expression if both are expressed concomitantly. The role of GATA3 in the regulation of ER is a continued avenue of research in our laboratory. Interestingly, we found that silencing of either HuR or GATA3 reduced cell viability to the same extent. No further decrease occurred if both were silenced together. As HuR controls GATA3 mRNA stability, this data indicates that GATA3 contributes to cell growth and suggests that reducing GATA3, either directly or indirectly through silencing of HuR, may be an effective mechanism to inhibit tumor cell growth.
Abbreviations
- AZA:
-
5-aza 2′ Deoxycytidine
- DNMT:
-
DNA methyltransferase
- ER:
-
Estrogen receptor
- HDAC:
-
Histone deacetylase
- TSA:
-
Trichostatin A
- WT:
-
Wild type
References
Mehra R, Varambally S, Ding L, Shen R, Sabel M, Ghosh D, Chinnaiyan A (2005) Kleer C: identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res 65:11259–11264
Ciocca V, Daskalakis C, Ciocca RM, Ruiz-Orrico A, Palazzo JP (2009) The significance of GATA3 expression in breast cancer: a 10-year follow-up study. Hum Pathol 40(4):489–495
Bertucci F, Houlgatte R, Benziane A, Granjeaud S, Adelaide J, Tagett R, Loriod B, Jacquemier J, Veins P, Jordan B et al (2000) Gene expression profiling of primary breast carcinomas using arrays of candidate genes. Hum Mol Genet 9(20):2981–2991
Hoch R, Thompson D, Baker R, Weigel R (1999) GATA-3 expression is association with estrogen receptor in breast cancer. Int J of Cancer 84:122–128
Shen D, Chang H, Chen Z, He J, Lonsberry V, Elshimali Y, Chia D, Seligson S, Goodglick L, Nelson S et al (2005) Loss of annexin A1 expression in human breast cancer detected by multiple high-throughput analyses. Biochem Biophys Res Commun 326:218–227
Usary J, Llaca V, Karaca G, Presswala S, Karaca M, He X, Langerod A, Karesen R, Oh D, Dressler L et al (2004) Mutation of GATA3 in human breast tumors. Oncogene 23:7669–7678
Wilson B, Giguere V (2008) Meta-analysis of human cancer microarrays reveals GATA3 is integral to the estrogen receptor alpha pathway. Mol Cancer 7(1):49
Asselin-Labat M-L, Sutherland K, Barker H, Thomas R, Shackleton M, Forrest N, Hartley L, Robb L, Grosveld F, van der Wees J et al (2007) Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol 9:201–209
Kouros-Mehr H, Bechis S, Slorach E, Littlepage L, Egeblad M, Ewald A, Pai S, Ho I, Werb Z (2008) GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 13:141–152
Kouros-Mehr H, Slorach E, Sternlicht M, Werb Z (2006) GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127:1041–1055
Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M (2007) Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res 67(13):6477–6483
Keen JC, Davidson NE (2003) The biology of breast carcinoma. Cancer 97(3 Suppl):825–833
Kenealy MR, Flouriot G, Pope C, Gannon F (1996) The 3′untranslated region of the human estrogen receptor gene post- transcriptionally reduces mRNA levels. Biochem Soc Trans 24(1):107S
Kenealy M-R, Flouriot G, Sonntag-Buck V, Dandekar T, Brand H, Gannon F (2000) The 3′-untranslated region of the human estrogen receptor alpha gene mediates rapid messenger ribonucleic acid turnover. Endocrinology 141(8):2805–2813
Ing NH, Massuto DA, Jaeger LA (2007) Estradiol up-regulates A+ U-rich RNA-binding factor 1 p45 binding to stabilizing regions within the 3′untranslated region of estrogen receptor alpha Mrna. J Biol Chem 283(3):1764–1772
Hostetter C, Licata LA, Witkiewicz A, Costantino C, Yeo CJ, Brody JR, Keen JC (2008) Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells. Cancer Biol Ther 7(9):1496–1506
Pryzbylkowski P, Obajimi O, Keen J (2008) Trichostatin A and 5 Aza-2′ deoxycytidine decrease estrogen receptor mRNA stability in ER positive MCF7 cells through modulation of HuR. Breast Cancer Res Treat 111:15–25
Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE (2001) Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res 61:7025–7029
Sharma D, Saxena NK, Davidson NE, Vertino PM (2006) Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated er recruits distinctive corepressor complexes. Cancer Res 66(12):6370–6378
Sharma D, Blum J, Yang X, Beaulieu N, Macleod A, Davidson NE (2005) Release of methyl CpG binding proteins and histone deacetylase 1 from the estrogen receptor alpha promoter upon reactivation in ER negative human breast cancer cells. Mol Endocrinol 19(7):1740–1751
Macaluso M, Cinti C, Russo G, Russo A, Giordano A (2003) pRb2/p130–E2F4/5-HDAC1-SUV39H1–p300 and pRb2/p130–E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor alpha in breast cancer. Oncogene 22:3511–3517
Macaluso M, Montanari M, Noto PB, Gregorio V, Bronner C, Giordano A (2007) Epigenetic modulation of estrogen receptor-{alpha} by pRb family proteins: a novel mechanism in breast cancer. Cancer Res 67(16):7731–7737
Adams BD, Furneaux H, White BA (2007) The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-{alpha} (ER{alpha}) and represses ER{alpha} messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol 21(5):1132–1147
Liu W–H, Yeh S–H, Lu C–C, Yu S-L, Chen H-Y, Lin C-Y, Chen D-S, Chen P-J (2009) MicroRNA-18a prevents estrogen receptor-[alpha] expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology 136(2):683–693
Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, Jacob S, Majumder S (2008) MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem 283(44):29897–29903
Zhao J-J, Lin J, Yang H, Kong W, He L, Ma X, Coppola D, Cheng JQ (2008) MicroRNA-221/222 negatively regulates estrogen receptor{alpha} and is associated with tamoxifen resistance in breast cancer. J Biol Chem 283(45):31079–31086
Keen JC, Sholl L, Wills-Karp M, Georas SN (2001) Preferential activation of nuclear factor of activated T cells c correlates with mouse strain susceptibility to allergic responses and Interleukin-4 gene expression. Am J Respir Cell Mol Biol 24(1):58–65
Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, Davidson NE (2003) A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor Î ± (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res Treat 81(3):177–186
Ferguson AT, Lapidus R, Baylin S, Davidson NE (1995) Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res 55:2279–2283
Ferguson AT, Vertino P, Spitzner J, Baylin S, Muller M, Davidson NE (1997) Role of estrogen receptor gene demethylation and DNA methyltransferase-DNA adduct formation in 5-aza-2′-deoxycytidine-induced cytotoxicity in human breast cancer cells. J Biol Chem 272(51):32260–32266
Yan L, Nass SJ, Smith D, Nelson WG, Herman JG, Davidson NE (2003) Specific inhibition of DNMT1 by antisense oligonucleotides induces re-expression of estrogen receptor a (ER) in ER-negative human breast cancer cell lines. Cancer Biol Ther 2(5):552–556
Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, Herman JG, Davidson NE (2000) Transcriptional activation of estrogen receptor α in human breast cancer cells by histone deacetylase inhibition. Cancer Res 60(24):6890–6894
Li L-C, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18(11):1427–1431
Hinman M, Lou H (2008) Diverse molecular functions of Hu proteins. Cell Mol Life Sci 65(20):3168–3181
Acknowledgments
The authors would like to thank Dr. Cristiana Stellato for her discussions with this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors Lauren A. Licata and Christine L. Hostetter are equally contributed to this work.
Rights and permissions
About this article
Cite this article
Licata, L.A., Hostetter, C.L., Crismale, J. et al. The RNA-binding protein HuR regulates GATA3 mRNA stability in human breast cancer cell lines. Breast Cancer Res Treat 122, 55–63 (2010). https://doi.org/10.1007/s10549-009-0517-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-009-0517-8