
Abstract
Purpose Dermatologic events (DEs) in patients with cancer treated with lapatinib, a small-molecule dual tyrosine kinase inhibitor (TKI) of epidermal growth factor receptor (EGFR [ErbB1]) and HER2 (ErbB2), were characterized. Patients and methods Nine clinical trials of metastatic cancer were included in this analysis. Lapatinib was administered at doses ranging from 1000 to 1500 mg/day as monotherapy (n = 928) or in combination with paclitaxel or capecitabine (n = 491). Patients not treated with lapatinib comprised the control group. Dermatologic events included hand-foot syndrome, rash, hair disorder, dry skin, pruritus/urticaria, skin disorder, skin infection, and nail disorder; DEs were characterized based on type, time to onset, severity, duration, and required interventions. Results Fifty-eight percent of patients treated with lapatinib monotherapy, 74% treated with lapatinib plus paclitaxel or capecitabine, and 53% in the control group developed DEs. Among patients receiving lapatinib monotherapy, 55% experienced grade 1/2 DEs, 3% had grade 3 DEs, and no grade 4 DEs were observed. The most common DE was rash (43%); all other events occurred in ≤8% of patients. Most DEs developed between days 1 and 14 of starting treatment, with a median duration of 29 days. Three percent of DEs led to lapatinib dose reduction, 7% resulted in dose interruption, and 1% led to drug discontinuation. Conclusions Most DEs in lapatinib-treated patients present early, are mild to moderate in severity, and infrequently require dose modification or treatment interruption. Lapatinib-associated DEs appear to differ clinically from those associated with EGFR TKIs in both frequency and severity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Epidermal growth factor receptor (EGFR) is a member of the ErbB family of transmembrane tyrosine kinase receptors that play a pivotal role in cell growth and survival, and overexpression of EGFR in tumors is associated with poor prognosis [1, 2]. Several EGFR inhibitors have been developed as anticancer agents including monoclonal antibodies (e.g., cetuximab and panitumumab) and small-molecule tyrosine kinase inhibitors (TKI; e.g., erlotinib and gefitinib). Epidermal growth factor receptor inhibitors are associated with specific dermatologic reactions, including a papulopustular rash that affects primarily the face and upper trunk, itchy skin (pruritus), inflammation around the nails (paronychia), xerosis, alopecia, and increased growth of the eyelashes and facial hair [3–6]. These dermatologic reactions reflect EGFR inhibition in epidermal and follicular keratinocytes, resulting in alterations in keratinocyte proliferation, differentiation, migration, and attachment [4]. Although rarely life-threatening, the physical and psychosocial distress associated with these dermatologic reactions may reduce compliance with EGFR inhibitors [7].
Lapatinib (Tykerb®, GlaxoSmithKline), an orally active small-molecule dual TKI of EGFR and HER2, is approved in several countries for the treatment of patients with recurrent HER2-positive advanced or metastatic breast cancer [8]. Lapatinib is also being investigated for the treatment of a variety of other solid tumors as mono- or combination therapy [9–12]. In a phase III study, lapatinib combined with capecitabine produced a 51% reduction in the risk of disease progression compared with capecitabine monotherapy without an increase in serious toxic effects or symptomatic cardiac events in patients with normal left ventricular ejection fraction at baseline [13]. In several studies, rash and diarrhea were identified as the most common side effects of lapatinib [14–16]. Unlike other TKIs where the occurrence and severity of rash appeared to correlate with plasma concentrations and clinical response, no such relationships were demonstrated for lapatinib [17]. This article reviews dermatology-related safety and tolerability data based on an analysis of nine phase II and III studies in patients with advanced/metastatic breast or non-breast cancers receiving lapatinib as mono- or combination therapy. Effective strategies that may be employed to manage lapatinib-associated dermatologic toxicity are discussed.
Patients and methods
Lapatinib clinical studies
Patients with advanced or metastatic cancer participated in this analysis of nine phase II or phase III clinical studies (Table 1). Five studies enrolled patients with breast cancer, and one study each enrolled patients with colorectal, non-small cell lung, bladder, and renal cancer. Lapatinib was used as monotherapy in seven studies and in combination with paclitaxel or capecitabine in two breast cancer studies. Lapatinib was administered once daily (QD) by mouth at 1000–1500 mg/day. Paclitaxel was administered intravenously (175 mg/m2 every 3 weeks) and capecitabine was administered as an oral medication (2000–2500 mg/m2 on days 1 through 14 of a 21-day treatment cycle). Ethics approval was obtained from the relevant local ethics committees, and the studies were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Patient assessments
Each patient’s medical history was recorded at the start of the study. If a patient had a dermatologic event (DE), the investigator would record and grade the event. Dose adjustments and modifications occurred on the basis of the physician’s decision and in consultation with the sponsor’s medical monitor. Dermatologic events that occurred in patients receiving study treatment were recorded during study visits. Patients with DEs underwent follow-up examinations, usually on a monthly basis, until 1 month after event resolution.
Characterization of dermatologic events
To characterize DEs, the Medical Dictionary of Regulatory Activities (MedDRA) was reviewed to identify searchable terms related to the System Organ Class of skin and subcutaneous disorders. Based on this review, a dermatology specialist identified eight categories of searchable terms and assigned all terms to an appropriate category: hand-foot syndrome, rash, hair disorder, dry skin, pruritus/urticaria, skin disorder, skin infection, and nail disorder. Each category included a range of lower-level searchable terms. For example, the rash category included the following terms: dermatitis acneiform, eczema, photosensitivity reaction, rash maculopapular, rash pustular, and skin ulcer, among others. Safety results from each of the nine studies were reviewed using these searchable terms to identify patients who experienced DEs. Patients were categorized by type of DE category, with a priority given to patients receiving combination therapy with capecitabine. More than one DE was recorded for some patients. Severity of DEs was graded on a scale from 1 to 4 using the National Cancer Institute Common Toxicity Criteria (version 2.0) and the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0) [18, 19]. Dermatologic events were assigned possible causality to the study medication by the investigators. Dermatologic events were also characterized based on time to onset, severity, duration, and treatment intervention (including lapatinib dose adjustment, interruption, or discontinuation). Patients enrolled in the same studies and who were not treated with lapatinib but received placebo, paclitaxel, capecitabine, or hormonal agents served as a control group.
Assessment of dermatologic events occurrence by origin or descent
Dermatologic events were assessed in various racial groups (Asian, black, white, Hispanic, or other), based on incidence, severity, treatment interventions, and clinical outcomes.
Pharmacokinetic assessments
Pharmacokinetic data were obtained from patients receiving either 1500 mg QD or 500 mg twice daily (BID) of lapatinib as monotherapy in 3 of the 9 studies (EGF20008, EGF20009, and EGF20014). Blood samples were collected during a 24-h dosing interval at the week 4 study visit to determine steady-state plasma concentrations of lapatinib. Plasma concentrations were measured using a previously reported liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) method [20]. Cumulative frequency of DEs in relation to plasma concentrations was fit using a Weibull cumulative density function.
Data analysis
Data included in the pooled analysis were taken from the data sets of the nine phase II and phase III clinical studies.
Results
Patient population
A total of 2,093 patients with locally advanced or metastatic cancer participated in nine completed phase II and III lapatinib clinical trials (Table 2); this patient population included 1,413 patients with breast cancer and 680 patients with non-breast cancer. Patients were treated with lapatinib as monotherapy (n = 926) or in combination with paclitaxel or capecitabine (n = 491). Patients who were not exposed to lapatinib but received paclitaxel, capecitabine, or hormonal agents served as controls (n = 676).
Patient demographics
The median age of study participants was 55 years (range, 19–87 years). The majority (79%) of patients were female. Most (78%) study participants were white. Hispanic (9%), Asian (8%), black (3%), or other (1%) patients represented the remaining patient populations. A higher percentage of non-white patients participated in breast cancer (2–13%) versus non-breast cancer studies (<3%). Most (96%) patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, and approximately two thirds of patients had a Karnofsky performance status score of 90–100 at study entry.
Dermatologic events
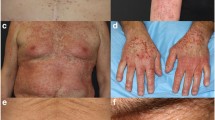
Dermatologic events were identified in 58% of patients who received lapatinib monotherapy, 70% of patients treated with lapatinib plus capecitabine, 76% of patients treated with lapatinib plus paclitaxel, and 53% of patients in the control group. Among patients treated with lapatinib monotherapy, DEs were reported as grade 1 or 2 in 55% of cases and grade 3 in 3% of cases, with no grade 4 DEs. Most (61%) DEs in patients treated with lapatinib combination therapy were grade 1 or 2. Grade 3 DEs occurred in 12% of patients. A total of 54% of lapatinib plus capecitabine-treated patients experienced grade 1 and 2 DEs, whereas 16% of patients experienced grade 3 events. Among lapatinib plus paclitaxel-treated patients, 66% experienced grade 1 and 2 events and 10% experienced grade 3 events. Skin rash (all grades, 43%; grade 3, 4%) was the most common DE in any lapatinib-treated patient (Table 3). Skin rash generally appeared on the trunk (Fig. 1); however, facial rash occurred in some patients. Hair disorder (3%), dry skin (3%), pruritus/urticaria (3%), skin disorder (1%), skin infection (<1%), and nail disorder (<1%) were among the less-frequently reported DEs. Among patients enrolled in the control arm of the nine clinical studies and treated with capecitabine monotherapy, the most common DE was hand-foot syndrome (all grades, 51%; grade 3, 14%). Patients in the control group who received paclitaxel monotherapy experienced rash as the most common DE (all grades, 24%; grade 3, <1%).

Examples of (a) skin rash (dermatitis) on back and (b) nail disorder (paronychia). The severity of dermatologic events was graded on a scale from 1 to 4 using the National Cancer Institute Common Toxicity/Terminology Criteria
Onset and duration of dermatologic events
Among the DEs described in Table 3, most developed early in the course of treatment; 44% of patients treated with any lapatinib combination therapy, 49% of patients treated with lapatinib monotherapy, and 36% of patients in the no-lapatinib (control) group developed DEs between days 1 and 14. Skin rash tended to be diagnosed early during both lapatinib treatment and no-lapatinib treatment (control group; approximately 50% each between days 1 and 14). The median duration was 29 days and 18 days for lapatinib-treated and control groups, respectively. Among lapatinib-treated patients, hand-foot syndrome was reported primarily with lapatinib plus capecitabine combination therapy and occurred at varying time points after drug initiation (34% between days 1 and 14, 30% between days 15 and 28, and 36% after 28 days), with a median duration of 22 days. Patients in the control group who received capecitabine monotherapy accounted for 43% of hand-foot syndrome cases between days 1 and 14, 23% between days 15 and 28, and 34% >28 days after drug initiation, with a median duration of 16 days.
Treatment intervention
Most (88%) DEs did not require lapatinib dose adjustment or treatment interruption (Table 4). Dose adjustments and treatment interruptions were more commonly reported in patients who received capecitabine monotherapy or lapatinib plus capecitabine. Among DEs requiring treatment intervention, 3% required dose adjustment, and 7% required treatment interruption. Only 1% of DEs resulted in treatment discontinuation.
Dermatologic events occurrence by origin or descent
The incidence of DEs was numerically higher in lapatinib-treated Hispanic patients (70%) compared with white (63%), black (62%), Asian (58%), or other (65%) patients. Among patients in the control group, the incidence of DEs was also higher in Hispanic patients (79% vs. 64% [black], 61% [Asian], 48% [white], or 83% [other]). The majority (>50%) of DEs in each racial group were grade 1/2. Grade 3 events occurred in <8% of lapatinib-treated patients. There were no major differences between racial groups in terms of treatment intervention; ≥85% of patients in each racial group did not require dose modification or treatment interruption. The percentage of DEs that resolved was lower in black patients (~50%) compared with any of the other racial groups (>70%).
Pharmacokinetic results
Plasma concentrations of lapatinib were available from 218 patients. Mean steady-state plasma concentrations were similar in patients receiving 1500 mg QD (1637 ng/ml) and 500 mg BID (1652 ng/ml) and were therefore combined. The range of plasma concentrations in patients with DEs (22–4576 ng/ml) was similar to that in patients with no DEs (25–4940 ng/ml), and there was no apparent relationship between the duration of DEs and lapatinib plasma concentrations. The 218 patients with plasma concentrations of lapatinib included 83 (38%) white, 67 (31%) Asian, 60 (27%) Hispanic, and 8 (4%) black patients. The incidence of DEs was similar to that reported in the larger subgroups in all studies for white (63%) and black patients (63%) but lower for Asian (34%) and Hispanic (37%) patients. The cumulative frequency of DEs in relation to plasma concentration indicated that more than 50% of patients experienced a DE at concentrations above 1793 ng/ml in white, 1932 ng/ml in black, 2068 ng/ml in Hispanic, and 2452 ng/ml in Asian patients (Fig. 2).

Cumulative frequency of dermatologic events in relation to lapatinib plasma concentrations for each ethnoracial subgroup
Discussion
This analysis of 2,093 patients with locally advanced or metastatic cancer demonstrated that DEs associated with lapatinib treatment were mild to moderate in severity in the majority of patients. Severe (grade 3) DEs occurred infrequently (6%), and there were no grade 4 DEs. Skin rash was the most common DE, occurring in 43% of lapatinib-treated patients (grade 3 in 4% of patients). Published prescribing information for erlotinib and sorafenib reported rash in 16–17% of placebo-treated patients with cancer [21, 22]. Therefore, rash would be reported in some patients whether or not the patient received the study medication.
Our analysis revealed that among patients not receiving lapatinib (capecitabine monotherapy, paclitaxel, or hormonal therapy), DEs were reported in 53% of patients compared with 58% of patients receiving lapatinib. Combination therapies influenced the development of DEs, with lapatinib plus capecitabine or paclitaxel-associated events reported in 74% of patients versus lapatinib monotherapy events in 58% of patients. The most marked example, hand-foot syndrome, was reported in approximately half (54%) of lapatinib plus capecitabine-treated patients versus 51% of capecitabine-monotherapy patients and <1% of lapatinib-monotherapy patients.
Among 2,093 patients, 2,319 DEs were reported. Dermatologic events tended to present early in the course of treatment, were usually of limited duration (median 29 days), were grade 1 or 2, and rarely required lapatinib dose adjustment or interruption. Among lapatinib-treated patients, dose modification occurred in approximately 10% of all events, and withdrawal resulting from DEs occurred in 1% of events. Dose modifications were primarily because of hand-foot syndrome (6%; 135/2,319 events), followed by skin rash (4%; 98/2,319 events), dry skin (<1%; 2/2,319 events), pruritus/urticaria (<1%; 1/2,319 events), and skin disorder (<1%; 1/2,319 events).
The occurrence of DEs in patients treated with lapatinib is thought to involve inhibition of EGFR in keratinocytes [23]. Lapatinib inhibits EGFR in vitro with an IC90 of 27 nM or 15.7 ng/ml [24]. Assuming in vivo inhibition is related to pharmacologically active unbound drug (<1%), the corresponding total (bound and unbound) plasma concentration of lapatinib would be at least 1570 ng/ml. This is consistent with lapatinib concentrations associated with an increased frequency of DEs (Fig. 2). However, plasma concentrations span similar ranges in patients with and without DEs, implicating other determinants such as environmental or genetic factors. The lower incidence of DEs observed in Asian patents may reflect the reported higher frequency of a dinucleotide repeat polymorphism in Intron 1 of the EGFR gene that leads to reduced transcription and expression of this protein [25]. The additional observation that the frequency of DEs in Asian patients was associated with higher lapatinib concentrations in contrast to white patients (2452 vs. 1793 ng/ml; p < 0.05) may reflect lower sensitivity to EGFR inhibition. A similar but smaller contrast was observed in Hispanics (2068 vs. 1793 ng/ml; p < 0.05), although the frequency of DEs was higher in the larger subgroup from all studies. Information on genetic or environmental factors that might distinguish this population is lacking.
Lapatinib is extensively metabolized and excreted in the form of several metabolites, only one of which is capable of inhibiting EGFR. However, this metabolite is less potent than the parent drug, unmeasurable in plasma, and accounts for only 8% of the excreted dose; therefore, this metabolite cannot achieve concentrations capable of inhibiting EGFR.
The EGFR is primarily expressed in undifferentiated, proliferating keratinocytes that are located in the basal and suprabasal layers of the epidermis and the outer root sheath of the hair follicle [26]. Drug-induced inhibition of EGFR affects keratinocytes by inducing growth arrest and apoptosis, decreasing cell migration, increasing cell attachment and differentiation, and stimulating inflammation, all of which result in distinct dermatologic manifestations [27]. Subsequently, inflammatory cells release chemoattractants that result in leukocyte recruitment and the release of enzymes linked to keratinocyte apoptosis and tissue damage [28]. Eventually, the thickness of the epidermis decreases and a thin stratum corneum with abnormal differentiation patterns develops, leading to latter DEs (e.g., xerosis).
Dermatologic events have also been reported in patients treated with other EGFR inhibitors. However, differences in the definition and grading of DEs make it difficult to compare the incidence and severity of DEs associated with different EGFR inhibitors [6]. Nevertheless, DEs appear to occur more frequently and with greater severity in patients treated with cetuximab, panitumumab, erlotinib, and gefitinib compared with lapatinib. For example, among patients with colorectal cancer who were treated with cetuximab monotherapy, the most common DEs included acneiform rash (90%), nail disorder (16%), and pruritus (11%) [29]. Dermatologic events in patients with colorectal cancer who were treated with panitumumab included erythema (65%), acneiform dermatitis (57%), pruritus (57%), rash (22%), and dry skin (10%) [30]. In patients with non-small cell lung cancer who were treated with erlotinib, rash (75%), pruritus (13%), and dry skin (12%) were among the most prevalent DEs [21]. Similarly, rash (43%), acne (25%), dry skin (13%), and pruritus (8%) were the most commonly reported DEs in gefitinib-treated non-small cell lung cancer patients [31]. Dermatologic events in patients treated with other EGFR inhibitors tended to occur within the first few weeks of therapy, and 9–17% of patients required dose reduction or treatment discontinuation.
Lapatinib-associated DEs appear to be clinically different from those associated with single-targeted EGFR agents (e.g., cetuximab, panitumumab, and erlotinib). For example, rash associated with lapatinib tends to be localized most frequently on the trunk and infrequently on the face, with pruritus being rare (3%), whereas rash occurred on the face in 82% of patients treated with gefitinib and cetuximab [32]. Hand-foot syndrome occurred at a lower incidence in the lapatinib group compared with the control group; this result is expected because some of the patients in the control group were treated with capecitabine, an agent that is associated with a high incidence of hand-foot syndrome (all grade, 54–63%) [33]. In a previous report, hand-foot syndrome occurred in half of the patients treated with capecitabine and the incidence of hand-foot syndrome was not increased by addition of lapatinib to the treatment regimen [13]. Rare occurrences of nail changes were challenging complications.
Few controlled clinical studies have investigated treatment options for EGFR inhibitor-associated dermatologic toxicities [34]; consequently, evidence-based treatment recommendations are scarce. A proactive approach, with patient education and early and frequent follow-up, is critical in managing lapatinib-associated cutaneous reactions. On initiation of lapatinib therapy, patients are advised to moisturize dry areas of the skin BID with a thick alcohol-free emollient. Patients should minimize exposure to sunlight and use a broad-spectrum sunscreen with a sun protection factor ≥30. Physical sunscreens, containing zinc oxide or titanium dioxide, are preferred over chemical sunscreens and should be applied 1–2 h before sun exposure and repeated if sun exposure is prolonged. Treatment intervention is usually based on the type and severity of the lapatinib-associated toxicity. For rash, oral steroids may be used for a short (maximum of 14 days) treatment course to help patients remain on study therapy. Topical or systemic agents (e.g., antihistamines or pregabalin) may be of benefit in the treatment of pruritic reactions [35]. Antiseptic baths, local potent corticosteroids (e.g., clobetasol ointment), and silver nitrate applications are recommended for paronychia. Culture-driven, topical, or systemic antibiotics are indicated for superinfected lesions. A dermatology consultation is encouraged for patients with extensive or symptomatic grade 3 or 4 DEs; chronic, persistent, or recurring lower-grade DEs; and in cases where physicians require another opinion regarding the treatment course of action.
Lapatinib treatment may be interrupted for up to 14 days in patients with grade 3 or 4 dermatologic reactions. Lapatinib may also be interrupted for grade 2 dermatologic reactions that do not improve after 2 weeks of therapy and significantly decrease the patient’s quality of life (Fig. 3). Rechallenge at the same dose may be considered for patients with grade 3 or 4 DEs that recover to grade 1 or 2 within 14 days after interrupting lapatinib therapy. Lapatinib treatment should be permanently discontinued if a grade 3 or 4 dermatologic reaction is intolerable to the patient despite recommended treatment interventions or when a grade 3 or 4 reaction is intolerable to the patient and recurs after drug interruption and rechallenge. Treatment should be immediately and permanently discontinued in the event of anaphylaxis or Stevens-Johnson’s Syndrome/toxic epidermal necrolysis.

Algorithm for the management of lapatinib-associated skin rash. BSA: body surface area; ADL: activities of daily living. Topical corticosteroids: hydrocortisone 2.5% or aclometasone 0.05% creams, bid. Oral semisynthetic tetracyclines: doxycycline or minocycline 100 mg bid
In conclusion, the majority of DEs in lapatinib-treated patients are mild to moderate in severity and of limited duration. Most DEs do not require lapatinib dose adjustments and are managed with established standard therapeutic regimens used to treat similar dermatologic conditions. Proactive management of DEs is recommended when lapatinib is combined with chemotherapy, and interventions should be tailored toward the toxicity and causal agent. Increased education and attention to dermatologic health will help health care professionals to view lapatinib-associated DEs as manageable and allow patients the clinical benefit gained from uninterrupted lapatinib therapy.
References
Dei Tos AP, Ellis I (2005) Assessing epidermal growth factor receptor expression in tumours: what is the value of current test methods? Eur J Cancer 41:1383–1392
Itakura Y, Sasano H, Shiga C et al (1994) Epidermal growth factor receptor overexpression in esophageal carcinoma. An immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer 74:795–804
Agero AL, Dusza SW, Benvenuto-Andrade C et al (2006) Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol 55:657–670
Lacouture ME (2006) Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer 6:803–812
Galimont-Collen AF, Vos LE, Lavrijsen AP et al (2007) Classification and management of skin, hair, nail and mucosal side-effects of epidermal growth factor receptor (EGFR) inhibitors. Eur J Cancer 43:845–851
Perez-Soler R, Saltz L (2005) Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol 23:5235–5246
Wagner LI, Lacouture ME (2007) Dermatologic toxicities associated with EGFR inhibitors: the clinical psychologist’s perspective. Oncology 21(11 suppl 5):34–36
Tykerb [package insert] (2007) GlaxoSmithKline, Research Triangle Park, NC
Fields AL, Rinaldi DA, Henderson CA et al (2005) An open-label multicenter phase II study of oral lapatinib (GW572016) as single agent, second-line therapy in patients with metastatic colorectal cancer [abstract]. J Clin Oncol 23(suppl):266s. Abstract 3583
Ross HJ, Blumenschein GR, Dowlati A et al (2005) Preliminary safety results of a phase II trial comparing two schedules of lapatinib (GW572016) as first line therapy for advanced or metastatic non-small cell lung cancer [abstract]. J Clin Oncol 23(suppl):645s. Abstract 7099
Wulfing C, Machiels J, Richel D et al (2005) A single arm, multicenter, open-label, phase II study of lapatinib as 2L treatment of patients with locally advanced/metastatic transitional cell carcinoma (TCC) of the urothelial tract [abstract]. J Clin Oncol 23(suppl):401s. Abstract 4594
Ravaud A, Gardner J, Hawkins R et al (2006) Efficacy of lapatinib in patients with high tumor EGFR expression: results of a phase III trial in advanced renal cell carcinoma (RCC). J Clin Oncol 24(suppl):217s. Abstract 4502
Geyer CE, Forster J, Lindquist D et al (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355:2733–2743
Dhillon S, Wagstaff AJ (2007) Lapatinib. Drugs 67:2101–2108; discussion 2109–2110
Mukherjee A, Dhadda AS, Shehata M et al (2007) Lapatinib: a tyrosine kinase inhibitor with a clinical role in breast cancer. Expert Opin Pharmacother 8:2189–2204
Montemurro F, Valabrega G, Aglietta M (2007) Lapatinib: a dual inhibitor of EGFR and HER2 tyrosine kinase activity. Expert Opin Biol Ther 7:257–268
Burris HA, Hurwitz HI, Dees EC et al (2005) Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol 23:5305–5313
Cancer Therapy Evaluation Program (1999) Common toxicity criteria, version 2.0. http://ctep.cancer.gov/forms/CTCv20_4-30-992.pdf. Accessed 7 Feb 2008
Cancer Therapy Evaluation Program (2006) Common terminology criteria for adverse events, version 3.0 (CTCAE). http://ctep.cancer.gov/forms/CTCAEv3.pdf. Accessed 7 Feb 2008
Hsieh S, Tobien T, Koch K et al (2004) Increasing throughput of parallel on-line extraction liquid chromatography/electrospray ionization tandem mass spectrometry system for GLP quantitative bioanalysis in drug development. Rapid Commun Mass Spectrom 18:285–292
Tarceva [package insert] (2007) OSI Pharmaceuticals Inc, Melville, NY
Nexavar [package insert] (2007) Bayer Pharmaceuticals Corp, West Haven, CT
Lynch TJ Jr, Kim ES, Eaby B et al (2007) Epidermal growth factor receptor inhibitor-associated cutaneous toxicities: an evolving paradigm in clinical management. Oncologist 12:610–621
Wood ER, Truesdale AT, McDonald OB et al (2004) A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res 64:6652–6659
Amador ML, Oppenheimer D, Perea S et al (2004) An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res 64:9139–9143
Nanney LB, Stoscheck CM, King LE Jr et al (1990) Immunolocalization of epidermal growth factor receptors in normal developing human skin. J Invest Dermatol 94:742–748
Kari C, Chan TO, Rocha de Quadros M et al (2003) Targeting the epidermal growth factor receptor in cancer: apoptosis takes center stage. Cancer Res 63:1–5
Lacouture ME, Lai SE (2006) The PRIDE (Papulopustules and/or paronychia, Regulatory abnormalities of hair growth, Itching, and Dryness due to Epidermal growth factor receptor inhibitors) syndrome. Br J Dermatol 155:852–854
Erbitux [package insert] (2007) Imclone Systems Inc, Branchburg, NJ
Vectibix [package insert] (2007) Amgen Inc, Thousand Oaks, CA
Iressa [package insert] (2005) AstraZeneca Pharmaceuticals, Wilmington, DE
Jacot W, Bessis D, Jorda E et al (2004) Acneiform eruption induced by epidermal growth factor receptor inhibitors in patients with solid tumours. Br J Dermatol 151:238–241
Xeloda [package insert] (2006) Roche Pharmaceuticals, Nutley, NJ
Jatoi A, Rowland K, Sloan JA et al (2007) Does tetracycline prevent/palliate epidermal growth factor receptor (EGFR) inhibitor-induced rash? A phase III trial from the North Central Cancer Treatment Group (N03CB) [abstract]. J Clin Oncol 25(suppl):494s. Abstract LBA9006
Lacouture ME, Cotliar J, Mitchell EP (2007) Clinical management of EGFRI dermatologic toxicities: US perspective. Oncology (Williston Park) 21:17–21
Acknowledgments
We would like to thank the patients who enrolled in these studies, the medical personnel who cared for them, and Shradha Sainju from GlaxoSmithKline. We would also like to thank Jeff Riegel, PhD, and Ann Marie Fitzmaurice, PhD, ProEd Communications, Inc.® for their medical editorial assistance. Dr. Lacouture is supported by a Zell Scholarship from the Robert H Lurie Comprehensive Cancer Center of Northwestern University. GlaxoSmithKline conducted the clinical studies (i.e., funded, designed, and analyzed data) that were included in the pooled analysis of skin events associated with lapatinib treatment. GlaxoSmithKline also provided statistical support for the pooled analysis and medical writing support for manuscript development. M.E. Lacouture and M. Koehler were involved in the conception of the manuscript. A.J. Preston, M. Koehler, M.E. Lacouture, R. Sweetman, and K.L. Blackwell contributed to the design of the analysis. S.M. Laabs, V.M. Salazar, M. Koehler, R.W. Sweetman, A. Di Leo, and H.L. Gomez were involved in patient recruitment. M.E. Lacouture, S.M. Laabs, M. Koehler, R.W. Sweetman, A. Di Leo, V.M. Salazar, and K.M. Koch were involved in the collection and assembly of data and in data analysis and interpretation. As safety manager for lapatinib, J.A. Byrne contributed data and interpretation of side effects. M.E. Lacouture, S.M. Laabs, M. Koehler, V.M. Salazar, K.M. Koch, K.L. Blackwell, and J.A. Byrne contributed to manuscript development. All authors reviewed, revised, and approved the final draft of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lacouture, M.E., Laabs, S.M., Koehler, M. et al. Analysis of dermatologic events in patients with cancer treated with lapatinib. Breast Cancer Res Treat 114, 485–493 (2009). https://doi.org/10.1007/s10549-008-0020-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-008-0020-7