
Abstract
Purpose Evaluation of the safety and efficacy of a combination of docetaxel and doxorubicin in breast cancer patients. Evaluation and comparison of the pathological complete response (pCR) to this regimen according to various definitions in different clinical trials. Utilize the data to propose standardization of definitions. Patients and Methods Between 1998 and 2001, 141 patients with stage II (tumor size >3.0 cm) or III breast cancer were treated with doxorubicin 50 mg/m2 followed by docetaxel 60 mg/m2 (AT) on day 1. A total of 4 courses of AT were administered as primary chemotherapy with intervals of 3 weeks. Additionally, 2 cycles of the same regimen were administered after surgery when clinical CR or PR was achieved; otherwise, 4 cycles of CMF were added postoperatively. Results 141 patients were enrolled in this trial. A clinical response rate was 86%. Seven patients (5%) achieved pCR according to the Japanese Breast Cancer Society classification, 14 patients (10%) fulfilled the University of Texas M.D. Anderson Cancer Center trial’s pCR criteria, and 19 patients (14%) met the NSABP trial pCR definition. NCI CTC version 2 grade 3/4 toxicities included leucopenia (26%), neutropenia (85%) and febrile neutropenia (12%). Conclusion Primary chemotherapy with AT induced modest tumor responses with tolerable toxicity. Differences in the definition of pCR among clinical trials caused substantial confusion in interpreting the trial results. Therefore, standardization of the pCR definition after primary chemotherapy is needed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is now widely accepted that primary chemotherapy achieves high clinical response rates and allows conservative surgery in more patients with breast cancer without compromising the prolonged disease-free and overall survival rates that were achieved after postoperative chemotherapy [1–5]. Tumor shrinkage by the primary chemotherapy can be easily monitored clinically both by physicians and patients [6]. For physicians, continuation of treatment is reasonably determined based on efficacy. For patients, compliance with the scheduled courses of chemotherapy is increased because they, themselves, experience the efficacy, which helps them mentally to overcome the unpleasant adverse effects. The pathological response to the primary chemotherapy provides reliable prognostic information [7]. Patients with a pathological complete response (pCR) have significantly longer disease-free and overall survival as compared to patients in lower pathological response categories [1]. However, the definitions used for the evaluation of pCR vary among clinical trials. For example, the Japan Breast Cancer Society defines pCR as no remaining cancer cells, or necrotic or non-viable residual cancer cells [8], and the German Prospective Adriamycin–Docetaxel (GEPARDO) Trial defines pCR as no microscopic evidence of viable tumor cells in resected specimens [9, 10]. In contrast, the definition adopted by the National Surgical Adjuvant Breast and Bowel Project (NSABP) B 18 trial allows specimens with intraductal residual tumor cells [1]. The trial of the University of Texas M.D. Anderson Cancer Center (M.D. Anderson) pCR criteria requires not only complete response of the primary lesion but also the disappearance of axillary lymph node metastasis [11]. The differences in the definition of pCR among clinical trials cause substantial confusion in interpreting the trial results. Standardization of the definition of pCR after primary chemotherapy is, therefore, essential. We tested the clinical and pathological responses to the combination of two of the most active cytotoxic agents, Doxorubicin (A) and Docetaxel (T), in patients with stages II or III breast cancer and compared pathological responses using several definitions. These agent have shown no cross-resistance and have different toxicity profiles [12–14].
Patients and methods
Patient population
A patient had to meet the following inclusion criteria to be enrolled into the trial, which had been approved by the Institutional Ethics Review Committee: Breast cancer confirmed histologically with core needle biopsy or incisional biopsy specimen; stage II (tumor size ≥3.0 cm in largest diameter by palpation) or III using the 1997 International Union Against Cancer (UICC) classification system; less than 70-years-old; tumor estrogen receptor-negative or progesterone receptor-negative; Eastern Cooperative Oncology Group (ECOG) performance status ≤1; adequate hematological, renal, and hepatic functions (WBC count ≥4,000/mm3 or the absolute neutrophil count ≥2,000/mm3, platelet count ≥100,000/mm3, serum creatinine ≤1.5 mg/100 ml, AST ≤80 IU/l and total bilirubin ≤1.5 mg/100 ml); normal electrocardiography; and written informed consent.
Patients were excluded from participation for the following reasons: Previous treatment for breast cancer; previous cancer with a disease-free interval of less than 5 years or a second primary malignancy; active infection; psychiatric illness; cardiac disease or other significant illness that might influence the tolerability of the treatment; and pregnancy or breast-feeding.
Study treatment
The regimens used were doxorubicin 50 mg/m2 as a 1-h intravenous infusion, followed by docetaxel 60 mg/m2 as a 1-h infusion on day 1. A total of four courses were administered with intervals of 3 weeks between courses. All patients received premedication consisting of dexamethasone (8 mg) and granisetron hydrochloride (3 mg) before doxorubicin administration. Oral dexamethasone, 4 mg, was continued twice daily on day 2 and once daily on days 1 and 3 after chemotherapy. Antibiotic prophylaxis could be given to patients with febrile neutropenia during the first course. Additional antiemetic treatment could be prescribed if needed.
Patients who achieved clinical CR or PR to this regimen received 2 additional cycles of the same regimen after surgery. Patients with SD or PD received a mastectomy when the tumor was operable, followed by 4 cycles of CMF (cyclophosphamide 600 mg/m2, methotrexate 40 mg/m2 and 5FU 600 mg/m2, all intravenously, once every 3 weeks).
All patients who were tumor estrogen receptor-positive or progesterone receptor-positive received tamoxifen 20 mg a day for 5 years after chemotherapy.
Median follow-up time was 36 months (range, 5 to 60 months).
Dose modification
Therapy could be postponed for a maximum of 3 weeks if the WBC count was <3,000/mm3 or the absolute neutrophil count was <1,500/mm3, platelet count was <100,000/mm3, serum creatinine was >1.5 mg/100 ml and total bilirubin was >1.5 mg/100 ml. If patients did not recover from toxicity during this period, protocol treatment had to be discontinued, and surgery was recommended. During the treatment, if febrile neutropenia lasted for ≥5 days, the platelet count was ≤25,000/mm3, or any grade 3 or 4 (National Cancer Institute Common Toxicity Criteria (NCI CTC) version 2) nonhematological toxicity occurred—except for alopecia, nausea, and vomiting—the dose of docetaxel was reduced to 50 mg/m2 in the next cycle. These dose reduction adjustments were maintained during subsequent cycles.
Surgery and radiation therapy
After completion of chemotherapy and clinical assessment of response, patients underwent appropriate surgery according to the size and location of the primary tumor. If the tumor was still too large for breast-conserving surgery, mastectomy was recommended. If the tumor size allowed breast-conserving surgery, then the surgical margins had to be free of invasive or noninvasive breast cancer at a width of at least 1 mm; otherwise, repeat excision had to be performed.
All patients who underwent breast-conserving surgery or who were diagnosed as stage III at study entry received radiotherapy. Radiotherapy was delivered with a dose of 50 Gy in 25 fractions over 5 weeks using tangential fields to the chest wall or regional lymph nodes.
Trial assessments
The diagnosis was established with core needle biopsy or incisional biopsy specimens of the primary tumor. Nuclear grade was assessed by histopathological evaluation; estrogen and progesterone receptors and HER2 status were determined by immunohistochemistry. The pretreatment work-up included a complete history and physical examination, complete blood cell counts with differential and platelet counts, blood chemistry analysis, electrocardiography, chest radiography, breast computed tomography, abdominal ultrasonography, bone scan, bilateral mammography and breast ultrasonography. The size of the breast tumor and axillary nodal status was determined by palpation. In patients with multifocal or multicentric breast tumors, the lesion with the largest diameters was targeted for follow-up.
The clinical response to primary chemotherapy was classified with palpation by the following criteria: complete response (CR), a total resolution of the breast tumor based on physical examination; partial response (PR), a 50% or greater reduction of the product of the two largest perpendicular dimensions of the breast mass; progressive disease (PD), a 25% or greater increase in the size of the breast mass or the appearance of a new lesion; Stable disease (SD), not showing enough change in disease status to be defined as PR or PD.
The pathological response to primary chemotherapy was evaluated using definitions employed by five clinical trials. The Japanese pathological response criteria was characterized as grade 3 (pCR) when there was no cancer cells, or necrotic or non-viable residual cancer cells; grade 2, severe cellular injury or replacement of 2/3 or more of the cancer cells; grade 1b, severe cellular injury or replacement of 1/3 to 2/3 of the cancer cells; grade 1a, mild cellular injury in a portion of the cancer cells, or severe cellular injury or replacement of less than 1/3 of the cancer cells by fibroblasts, histiocytes or fibrosis; grade 0, no histological change in the cancer cells [8]. The Aberdeen classification characterized grade 1 as, some alteration to individual malignant cells but no reduction in overall number; grade 2, a minor loss of invasive tumor cells but overall cellularity still high; grade 3, a moderate reduction in tumor cells with up to an estimated 90% loss; grade 4, a marked disappearance of invasive cells such that only small clusters of widely dispersed cells are detected; grade 5, no invasive cells identifiable in sections from the site of the previous tumor, i.e., only in-situ disease or tumor stroma remained [15]. The GEPARDO classification characterized grades 0 and 1 as, “no effect” and “resorption and tumor sclerosis,” respectively; grade 2, minimal focal invasive tumor residues of less than 5 mm in diameter are found; grade 3, only in situ tumor residues are found; grade 4, not a single viable tumor cell could be detected [9]. In the NSABP B18 classification, pINV was defined as histological evidence of invasive cells among complete clinical responders; pCR, no histological evidence of invasive cells among complete clinical responders [1].
Although four definitions evaluate the primary lesion only, the M.D. Anderson trial’s pCR criteria also require the evaluation of axillary lymph nodes [11].
All patients who received at least one course of chemotherapy were assessable for toxicity. NCI CTC version 2 was used for evaluation.
Statistical considerations
The primary study end point was the clinical response rate. We estimated that a total sample size of 140 patients would be required to allow for 130 assessable patients. This sample size was required to demonstrate an anticipated clinical response rate of 80% with confidence intervals of 72–86% at an alpha of 0.05.
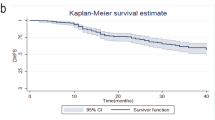
Recurrence-free survival was defined as time on study before local, regional, or distant tumor recurrence; second primary cancers, contralateral events, and deaths without evidence of disease were treated as censoring events. Patients who died without documented tumor recurrence were censored on the day of death or last follow-up. Patients who did not expire were censored at the time they were last known to be alive. The Kaplan–Meier curve [16] was used to estimate the recurrence-free survival of patients who entered this study.
Results
Between May 1998 and August 2001, 141 patients (median age, 49 years; range, 29–69 years) were enrolled in this study. The characteristics of the patients are summarized in Table 1. The median diameter of the largest primary tumor was 5.3 cm (3.0–12 cm), with 45% T2 tumors, 31% T3 tumors and 24% T4 tumors. A total of 92 patients had hormone receptor-positive tumors and 11 patients had HER2 overexpression. A total of 83% of tumors were invasive ductal carcinoma, whereas 5.6% were intraductal carcinoma.
Dose administration
Patients received a total of 560 cycles of doxorubicin and docetaxel. Chemotherapy was administered without dose reduction in 45 patients. The dose intensity of A and D were 94% and 76%, respectively.
Clinical response
A tumor response was assessed in 141 patients. The clinical response rate was 86% (95% Confidence Interval (CI): 78.9–91.1). Clinical CR was attained in 24 patients (17%), PR in 97 patients (69%), SD in 17 patients (12%) and PD in three patients (2%). Table 2 shows the number of responding patients during the chemotherapy. One PD patient underwent surgery after chemotherapy, two PD patients were not operable due to the extent of tumor progression. One patient who achieved PR refused surgical treatment.
Pathological response
The results of the pathological examination after chemotherapy were available for 138 patients. In seven patients (5%: 95%CI, 2.1–10.2), no tumor cells could be detected (the Japanese criteria, grade 3; the GEPARDO criteria, grade 4). In a further 12 patients (14%: 95%CI, 8.5–20.7), only intraductal cancer cells were found (defined as pCR in the NSABP 18 criteria and as pCR, grade 5 by the Aberdeen criteria), and in 25 patients, minimal focal invasive tumor residues of less than 5 mm in diameter were reported (defined as grade 2 in the GEPARDO criteria). A total of 14 patients (10%: 95%CI, 5.7–16.4) fulfilled the M.D. Anderson pCR criteria (Table 3). Thus, significant signs of pathological regression (grade 3 and grade 2 according to the Japanese criteria) of the primary tumor were reported in 49 patients (36%).
Rate of breast-conserving surgery
It was possible to conserve the affected breast in 31% of the patients. The rates of breast conservation were 40%, 27% and 12% in tumors with initial palpable sizes of ≥3 cm but less than 5 cm (n = 65), ≥5 cm but less than 7 cm (n = 55), and ≥7 cm (n = 17), respectively. The odds of conserving the breast were highly dependent on the initial palpable size of the tumor and the clinical response to primary chemotherapy.
Recurrence-free survival
Figure 1 shows the recurrence-free survival. The median follow-up time was 36 months (range, 5–60 months). The 3-year recurrence-free survival is 74.6%.

Kaplan–Meier analysis of recurrence-free survival time
Toxicity
All 141 patients who received at least one course of chemotherapy were assessable for toxicity. Table 4 shows the figures for NCI CTC version 2 grades 3/4 hematological and nonhematological toxicities. Leukopenia reached grade 3/4 in 26% of the patients, and grade 3/4 neutropenia was recorded in 85% of them. The incidence of febrile neutropenia was 12%. Only three patients were hospitalized due to this event. Other frequent toxicities, apart from alopecia, were nausea, vomiting, stomatitis and diarrhea.
Discussion
Breast cancer is widely regarded as a systemic disease [17, 18]. Preoperative administration of systemic therapy therefore represents a logical step in improving the efficacy of treatment. Many studies have reported that primary chemotherapy induced survival improvement that was comparable to that achieved with postoperative chemotherapy [19–21]. In addition, this therapy significantly improves the rate of breast-conserving surgery, and pCR serves as an important prognostic indicator [1, 7].
Newer regimens, including more active agents in primary chemotherapy, must have a potential to improve efficacy. In order to compare different primary chemotherapeutic regimens, it is important to use pCR as a primary endpoint in evaluating each regimen [22].
In defining pCR, some investigators evaluate it based on the state of the primary lesion alone, whereas others evaluate it based on the states of both the primary lesion and axillary lymph nodes; a consensus has not been reached among the clinical trial groups. For example, the NSABP B 18 trial regarded patients as pCR if their primary lesion had disappeared. Therefore, in some patients who were evaluated as having achieved pCR according to this criterion, metastatic foci remained in the axillary lymph nodes. Aberdeen’s pCR criteria, which is used at the University of Aberdeen (United Kingdom), is also based on this definition. In the M.D. Anderson Cancer Center trial, patients were evaluated as pCR if both the primary lesion and axillary metastasis had disappeared.
With respect to evaluation of the primary lesion, some investigators define pCR as the complete disappearance of cancer cells, whereas others allow the presence of intraductal residual tumor cells. Several kinds of criteria established in Japan and in Germany, GEPORDO, are based on the former definition of pCR, whereas other kinds of criteria established by the trials of NSABP B18, Aberdeen, and M.D. Anderson Cancer Center are based on the latter definition.
In addition, the criteria for preparing pathological specimens have not been standardized. Further investigations with thinner resected specimens may decrease the rate of pCR. In this study, we cut the primary lesion and its periphery to a thickness of 1.5 cm from surgical specimens in patients who underwent mastectomy, and cut surgical specimens to a thickness of 1 cm to prepare pathological specimens from patients who underwent breast-conserving surgery.
Among these criteria, those that appear to provide reasonably accurate predictions of the prognosis during long-term follow-up will be mainly employed among clinical trials in the future. At the present time it is not possible to tell which of these criteria should be preferably used because the follow-up periods have been too short. For an analysis of the criteria to use for obtaining the most accurate prognosis, a sufficient number of patients and thorough follow-up are needed; currently, only the NSABP B18 trial meets these conditions [23]. However, the NSABP criteria differ from the criteria used in Japan and Europe.
Thus, to summarize, the criteria for the pathological response to primary chemotherapy have not been standardized. Therefore, values should not be simply compared when the efficacy of chemotherapeutic regimens are evaluated based on the pCR rates.
In the future, primary chemotherapy will be performed in an increasing number of patients; therefore, standardized pathological criteria should be established.
References
Fisher B, Bryant J, Wolmark N et al (1998) Effect of preoperative chemotherapy on the outcome of women with operable breast cancer. J Clin Oncol 16:2672–2685
Bonadonna G, Valagussa P, Zucali R et al (1995) Primary chemotherapy in surgically resectable breast cancer. CA Cancer J Clin 45:227–243
Harris L, Swain SM (1996) The role of primary chemotherapy in early breast cancer. Semin Oncol 23:31–42
Smith IE, Walsh G, Jones A et al (1995) High complete remission rates with primary neoadjuvant infusional chemotherapy for large early breast cancer. J Clin Oncol 13:424–429
Anderson ED, Forrest AP, Hawkins RA et al (1991) Primary systemic therapy for operable breast cancer. Br J Cancer 63:561–566
Kaufmann M,Kubli F (1983) Current state of chemosensitivity testing of tumors. Dtsch Med Wochenschr 108:150–154
Kuerer HM, Newman LA, Smith TL et al (1999) Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J Clin Oncol 17:460–469
Kurosumi M, Akiyama F, Iwase T et al (2001) Histopathological criteria for assessment of therapeutic response in breast cancer. Breast Cancer 8:1–2
von Minckwitz G, Costa SD, Raab G et al (2001) Dose-dense doxorubicin, docetaxel, and granulocyte colony-stimulating factor support with or without tamoxifen as preoperative therapy in patients with operable carcinoma of the breast: a randomized, controlled, open phase IIb study. J Clin Oncol 19:3506–3515
Sinn HP, Schmid H, Junkermann H et al (1994) Histologic regression of breast cancer after primary (neoadjuvant) chemotherapy. Geburtshilfe Frauenheilkd 54:552–558
Green MC, Buzdar AU, Smith T et al (2005) Weekly paclitaxel improves pathologic complete remission in operable breast cancer when compared with paclitaxel once every 3 weeks. J Clin Oncol 23:5983–5992
von Minckwitz G, Costa SD, Eiermann W et al (1999) Maximized reduction of primary breast tumor size using preoperative chemotherapy with doxorubicin and docetaxel. J Clin Oncol 17:1999–2005
Ravdin PM, Burris HA 3rd, Cook G et al (1995) Phase II trial of docetaxel in advanced anthracycline-resistant or anthracenedione-resistant breast cancer. J Clin Oncol 13:2879–2885
Valero V, Holmes FA, Walters RS et al (1995) Phase II trial of docetaxel: a new, highly effective antineoplastic agent in the management of patients with anthracycline-resistant metastatic breast cancer. J Clin Oncol 13:2886–2894
Smith IC, Heys SD, Hutcheon AW et al (2002) Neoadjuvant chemotherapy in breast cancer: significantly enhanced response with docetaxel. J Clin Oncol 20:1456–1466
Kaplan E, Meire P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457–481
Bonadonna G, Valagussa P, Moliterni A et al (1995) Adjuvant cyclophosphamide, methotrexate, and fluorouracil in node-positive breast cancer: the results of 20 years of follow-up. N Engl J Med 332:901–906
Early Breast Cancer Trialist’ Collaborative Group (1998) Polychemotherapy for early breast cancer: an overview of the randomised trials. Lancet 352:930–942
van der Hage JA, van de Velde CJ, Julien JP et al (2001) Preoperative chemotherapy in primary operable breast cancer: results from the European Organization for Research and Treatment of Cancer trial 10902. J Clin Oncol 19:4224–4237
Makris A, Powles TJ, Ashley SE et al (1998) A reduction in the requirements for mastectomy in a randomized trial of neoadjuvant chemoendocrine therapy in primary breast cancer. Ann Oncol 9:1179–1184
Scholl SM, Fourquet A, Asselain B et al (1994) Neoadjuvant versus adjuvant chemotherapy in premenopausal patients with tumours considered too large for breast conserving surgery: preliminary results of a randomised trial: S6. Eur J Cancer 30A:645–652
von Minckwitz G, Raab G, Caputo A et al (2005) Doxorubicin with cyclophosphamide followed by docetaxel every 21 days compared with doxorubicin and docetaxel every 14 days as preoperative treatment in operable breast cancer: the GEPARDUO study of the German Breast Group. J Clin Oncol 23:2676–2685
Wolmark N, Wang J, Mamounas E et al (2001) Preoperative chemotherapy in patients with operable breast cancer: nine-year results from National Surgical Adjuvant Breast and Bowel Project B-18. J Natl Cancer Inst Monogr:96–102
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mukai, H., Watanabe, T., Ando, M. et al. Assessment of different criteria for the pathological complete response (pCR) to primary chemotherapy in breast cancer: standardization is needed. Breast Cancer Res Treat 113, 123–128 (2009). https://doi.org/10.1007/s10549-007-9889-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-007-9889-9