
Abstract
Estrogen receptor β gene codes for a variety of transcript isoforms resulting from alternative splicing, which are expressed both in mammary gland and in breast cancer cells. We studied the function of two exon-deleted ERβ isoforms recently identified by our group in comparison to ERβ1 in regulation of growth, apoptosis and gene expression of two breast cancer cell lines with different ERα status. Overexpression of ERβ1, but not of the exon-deleted variants exerted strong antitumoral effects both on ERα-positive MCF-7 and ERα-negative SK-BR-3 cells. ERβ1 overexpression slowed growth of MCF-7 and SK-BR-3 cells in the absence of E2 and also inhibited E2-triggered growth stimulation of MCF-7 cells, but overexpression of the exon-skipped variants did not affect cell growth. Whereas overexpression of ERβ1 triggered an increased basal and tamoxifen-induced apoptosis of MCF-7 and SK-BR-3 cells, the isoforms ERβδ125 or ERβδ1256 did not affect cellular tamoxifen response. The observed lack of function of the exon-deleted variants in terms of regulation of proliferation was accompanied both by their inability to affect expression of cyclins D1 and A2, p21 (WAF1) and PR and their disability to modulate estrogen response element (ERE) activation. In contrast, our results demonstrating antitumoral effects of ERβ1 on breast cancer cells with different ERα-status support the hypothesis that ERβ is able to exert antitumoral actions both on ERα-positive and -negative breast cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Estrogens are pivotal in the growth and development of both normal and neoplastic mammary tissues, and mediate most of their action via ligand-dependent transcription factors called estrogen receptors (ER). Despite the fact that ER status is already an important biomarker in breast cancer [1] situation is now appreciated to be far more complex. Identification of a second ER, called ERβ [2] has led to a re-evaluation of estrogen action in target tissues such as breast tumours. ERβ is clearly expressed in both normal and neoplastic human breast tissue [3, 4] although its role in either is unknown. In animal studies, while ERα has been shown to be essential for normal mammary gland development, ERβ effects are more subtle, with roles in terminal differentiation [5] and modulation of ERα activity being described [6–8]. In contrast to ERα, published data suggest that ERβ expression declines during breast tumorigenesis [3, 9]. This downregulation of ERβ in breast tumors compared with normal breast tissue suggests a role for ERβ as a tumor suppressor [10]. Nevertheless, ERβ expression in breast tumors varies widely [4, 11] and attempts to correlate ERβ with various biomarkers have resulted in varied, often contradictory conclusions [12]. This might also be due to differential detection of variant non-ligand binding ERβ proteins which have been detected in breast tissues [13, 14] and which code for proteins exerting functions distinct from that of the full-length ERβ1 protein [8, 15]. While the role of ERβ in breast cancer is unclear, one important currently emerging hypothesis is that increased expression of ERβ is associated with increased likelihood of response to endocrine therapy. Several studies report that increased levels of ERβ were associated with a better disease outcome and consistent with the breast tumour being tamoxifen sensitive [16–20].
However, when considering ERβ expression in breast cancers in vivo there are two groups, one where ERβ is coexpressed with ERα (ERβ+/ERα+) and the other where ERβ is expressed alone (ERβ+/ERα−). The first coexpressing group comprises ∼59% of primary human breast cancers [17, 21], while the ERβ alone expressing group comprises ∼17% of breast cancers [17, 21]. Emerging data support a differential function of ERβ when it is expressed alone compared with when it is co-expressed with ERα [22–25]. These observations might be explained by the fact that in coexpressing cells, the predominant mechanism of ERβ action might be negative modulation of ERα, whereas ERβ forms homodimers in cells not expressing ERα, thereby exerting alternative functions [6, 8, 26].
Another feature of both ERs is the variety of their mRNA isoforms resulting from differential splicing [27–31]. The so far identified ERβ splice variants are characterized by alternative 3′-exons (ERβ2, ERβ3, ERβ4, ERβ5) or by deletion of single or multiple exons (e.g. ERβδ2, ERβδ5/6). Some of these mRNA isoforms were demonstrated to code for ERβ proteins which are characterized by impaired estrogen or DNA binding or altered cofactor interaction [32, 33]. The emerging picture of multiple ERβ mRNA isoforms, and thus also the multitude of differentially built proteins, strongly suggests their synthesis to be considered as another level of complexity of estrogen signaling.
Previously we succeeded in molecular cloning of two novel ERβ splice variants from MDA-MB-231 breast cancer cells lacking exons 1, 2 and 5 or 1, 2, 5, and 6, respectively, which could be demonstrated to be expressed in a variety of human tissues [34]. In this study, we examined the function of these exon-deleted ERβ isoforms in comparison to ERβ1 in two breast cancer cell lines with different ERα status.
Materials and methods
Materials
Phenol red-free DMEM culture medium was obtained from Invitrogen (Karlsruhe, Germany), FCS was purchased from PAA (Pasching, Austria). 17-β estradiol, 4-OH tamoxifen, ICI 182,780, staurosporine and serum replacement 2 (SR2) were obtained from Sigma (Deisenhofen, Germany), MCF-7 and SK-BR-3 breast cancer cells were obtained from American Type Culture Collection (Manassas, USA). M-MLV-P reverse transcriptase, Cell Titer Blue kit, Caspase-Glo 3/7 kit and ImProm-II™ Reverse Transcriptase were purchased from Promega (Mannheim, Germany). RNeasy Mini Kit, RNase Free DNase Set and Quantitect SYBR Green PCR Kit were obtained from Qiagen (Hilden, Germany). PCR primers were synthesized at Metabion (Planegg-Martinsried, Germany). Transfectin reagent was obtained from BioRad (Hercules, USA). Platinum Pfx Polymerase and OptiMEM medium were purchased at Invitrogen (Karlsruhe, Germany). Rapid-Scan gene expression panel was obtained from Origene (Rockville, USA).
Plasmids
Vector pTARGET (Promega, Mannheim, Germany) allows cloning in E.coli and additionally carries the human cytomegalovirus (CMV) immediate-early enhancer/promoter region to promote constitutive expression of cloned DNA inserts in mammalian cells. This vector also contains the neomycin phosphotransferase gene, a selectable marker for mammalian cells. pTARGET derivatives containing ORFs of ERβ1, ERβδ125 or ERβδ1256 were used for overexpression in MCF-7 and SK-BR-3 cells. Vector pEGFP-N2 (Clontech) codes for the GFP protein for visualization of transfection efficacy using a fluorescence microscope. Vector pTAL-SEAP (Clontech) constitutively codes for the secreted alkaline phosphatase (SEAP) protein and served as positive control for the SEAP assay, and pTAL-ERE-SEAP is a reporter gene vector containing EREs in the promotor of the SEAP gene. Both vectors were used for the reporter gene assays performed in this study. Vector pSV-β-GAL (Promega) constitutively codes for the β-galactosidase enzyme and was used as internal control for transfection efficacy in the reporter gene assays.
Cell culture and transfections
MCF-7 and SK-BR-3 breast cancer cells were maintained in phenol red-free DMEM/F12 medium supplemented with 10% FCS or 1 × serum replacement (SR2). Cells were cultured with 5% CO2 at 37°C in a humidified incubator. For transfection, 4 × 105 cells per well of a 6-well dish were seeded in DMEM/F12 10% FCS. The next day, 2 ml fresh culture medium was added to the cells and transfection solution was prepared by mixing 5 μl Transfectin reagent (BioRad) and 1 μg plasmid DNA in OptiMEM reduced serum medium (Invitrogen) and added to the cultured cells. For generation of stable clones, G418 selection (300 μg/ml) was started 48 h after transfection.
Reverse transcription and PCR
Total RNA was isolated by means of the RNeasy kit (Qiagen) according to the manufacturer′s instructions. From 1 μg total RNA, cDNA was synthesized using 100 U M-MLV-P reverse transcriptase (Promega), 2.5 mM dNTP mixture and 50 pM random primers (Invitrogen). For detection of ERβ splice variants by standard RT-PCR, 2 μl of cDNA was amplified in a reaction mix of 1 U Platinum Polymerase (Invitrogen), 20 pmol of each primer, 1× PCR-buffer, 1.5 mM MgCl2 and 2.5 mM of each dNTP. The cDNA was amplified in 35 cycles (1 cycle = 1 min at 94°C melting, 2 min at 56°C annealing, 3 min at 72°C extension). All PCR primers were designed intron-spanning, sequences are indicated in Table 1, position of ERβ primers is illustrated in Fig. 1a.

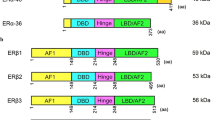
Characterization of the splice variants and breast cancer models used in this study. (a) mRNA and expected protein structure of the exon-deleted isoforms in comparison to ERβ1. Arrows indicate the position of PCR primers used in this study. UTR = untranslated region; AUG = translation initiation codon; DBD = DNA binding domain; LBD = ligand binding domain; AF-1/2 = activation function 1/2. (b) Expression of estrogen receptors in MCF-7 and SK-BR-3 breast cancer cells was determined by RT-PCR. MDA-MB-231 cells were used as a positive control for ERβ expression, β-actin as a positive control for cDNA integrity. (c) Relative transcript levels of ERβ1 and the exon-deletion variants in MCF-7 and SK-BR-3 breast cancer cells. In comparison to wildtype cells, the relative mRNA levels detected after overexpression of the respective ERβ variant are shown. Specific ERβ mRNA levels of clones isolated after G418 selection were determined by means of real time RT-PCR using a Light Cycler device (Roche, Germany) in comparison to samples which were not reversely transcribed as described in the materials and methods section and are expressed as % of the corresponding β-actin mRNA level (n = 3). (d) Estrogen response of MCF-7 and SK-BR-3 cells: activation of estrogen response elements (ERE) after stimulation with 1 nM 17β-estradiol for 24 h. ERE activation was determined by luminometric quantification of secreted SEAP protein by means of the Phospha Light Assay (Applied Biosystem). Cells were lysed using the Beta-Glo Assay (Promega) and subjected to this assay for luminometric determination of transfected β-galactosidase enzyme as internal control for the transfection efficacy. Both luminometric SEAP and β-GAL quantification were carried out using a VICTOR3 multilabel plate reader (PerkinElmer). To normalize the data, SEAP values are expressed in relation to the measured β-GAL values (n = 3). Data are expressed in % of the untreated (solvent EtOH) control. *P < 0.05 vs. untreated control
For real time PCR detection of ERβ isoforms or estrogen target genes (primer sequences in Table 1), 2 μl cDNA were amplified using the Quantitect SYBR Green PCR Kit (Qiagen) and the LightCyler PCR device (Roche Diagnostics, Mannheim, Germany). The PCR program was 95°C for 15 min, followed by 35 PCR cycles (95°C for 10 s, 56°C for 30 s, 72°C for 30 s) and a final extension for 5 min at 72°C, followed by a standard melting curve analysis. In all RT-PCR experiments, a 190 bp β-actin fragment was amplified as reference gene using intron-spanning primers actin-2573 and actin-2876. After performing dilution experiments with sample cDNA over a 100-fold range confirming the PCR efficiencies of all primer pairs to be approximately equal [35], data were analyzed using the comparative ΔΔCT method [36] calculating the difference between the threshold cycle (CT) values of the target and reference gene of each sample and then comparing the resulting Δ CT values between different samples. In these experiments, mRNA not subjected to reverse transcription was used as a negative control to distinguish cDNA and vector or genomic DNA amplification.
Cell viability assay
SK-BR-3 or MCF-7 wildtype (WT) cells or ERβ clones cultured in DMEM containing 1 × serum replacement 2 (SR2) were seeded in 96-well plates in triplicates (1000 cells/well), and were treated with 100 nM DPN, 100 nM PPT or 1 nM 17-β estradiol alone or in combination with 4-OH tamoxifen (100 nM). After 0, 72, 96 and 120 h, relative numbers of viable cells were measured in comparison to the untreated control and the solvent control using the fluorimetrical, resazurin-based Cell Titer Blue assay (Promega) according to the manufacturer’s instructions at 560Ex/590Em nm in a Victor3 multilabel counter (PerkinElmer, Germany). Cell growth was expressed as percentage of day 0 or percentage of the untreated medium control. Statistical analysis of the data was performed by one-way ANOVA using Prism 2.0 Software (Graph pad, San Diego, USA), with statistical significance accepted at P < 0.05.
Apoptosis assays
Wildtype cells or ERβ-clones cultured in DMEM supplemented with 1 × serum replacement 2 (SR2, Sigma) were seeded in 96-well plates (5000 cells/well) and treated with 1 nM 17-β estradiol in combination with different concentrations of 4-OH tamoxifen. After 6 h of treatment, cellular apoptosis was determined by measurement of caspase 3 and 7 activity by means of the luminometric Caspase-Glo 3/7 assay (Promega) according to the manufacturer’s protocol using a Victor3 multilabel counter (PerkinElmer, Germany). Additionally, apoptosis was measured by means of the Annexin V-FLUOS Staining Kit (Roche, Germany). Cells were treated with Annexin V and propidium iodide (PI) according to the manufacturer’s protocol, and apoptotic cells exhibiting positive green Annexin V fluorescence but no red PI staining were counted. Cellular apoptosis was expressed as percentage of the untreated control cells. Statistical analysis of the data was performed by one-way ANOVA using Prism 2.0 Software (Graph pad, San Diego, USA), with statistical significance accepted at P < 0.05.
Reporter gene assays
MCF-7 wildtype cells, ERβ-modulated MCF-7 clones and vector transfected MCF-7 control cells were seeded in 6-well plates in DMEM/F12 supplemented with 5% FCS (4 × 105 cell per well), five hours later serum concentration was reduced to 1% and 0.5 × serumfree SR2 medium was added. The next day, prior to transfection medium was changed to 1 × SR2. Transfections were carried out mixing 10 μl Transfectin reagent (BioRad, Hercules, USA) in a total volume of 250 μl OptiMEM medium with 5 μg pEGFP-N2 vector (Clontech) for easy visualization of transfection efficacy using a fluorescence microscope, 5 μg pTAL-SEAP vector (Clontech) as positive control for the SEAP assay, or 10 μg of reporter gene vector pTAL-ERE-SEAP (Clontech). Generally, 5 μg of the pSV-β-GAL vector (Promega) was added to the transfection solution serving as internal control for transfection efficacy. 24 h after adding the 250 μl transfection solution to the medium, cells were stimulated with 100 nM DPN, 100 nM PPT or 1 nM 17-β estradiol alone or in combination with 100 nM 4-OH tamoxifen or 100 nM ICI 182,780 in fresh DMEM/F12 containing 1 × SR2. The next day, medium was removed and 20 μl of it were subjected to the Phospha Light Assay (Applied Biosystem) for luminometric quantification of secreted SEAP protein in the culture supernatant according to the instructions of the manufacturer. Cells were lysed using the Beta-Glo Assay (Promega) and subjected to this assay for luminometric determination of transfected β-galactosidase enzyme as internal control for the transfection efficacy. Both luminometric SEAP and β-GAL quantification were carried out using a VICTOR3 multilabel plate reader (PerkinElmer). To normalize the data, SEAP values are expressed in relation to the measured β-GAL values.
Results
Overexpression of ERβδ125 or ERβδ1256 in breast cancer cell lines
Recently we identified two new ERβ splice isoforms lacking ERβ1 exons 1, 2 and 5 (termed ERβδ125) or exons 1, 2, 5 and 6 (termed ERβδ1256) from human MDA-MB-231 breast cancer cells, which are expressed in a variety of human tissues [34]. The deduced variant ERβ proteins are predicted to use an alternative translation start codon in exon 3 in the same reading frame as ERβ1, do not contain the AF-1 domain and have deletions both in ligand binding domain (LBD) and DNA binding domain (DBD) (Fig. 1a). Given that MCF-7 breast cancer cells express ERα but only small amounts of ERβ1 (Fig. 1b, c) and SK-BR-3 cells do not express notable amounts of both receptors (Fig. 1b, c), we first examined expression of ERβδ125 and ERβδ1256 in both cell lines by real time PCR. To confirm specificity of amplification of the exon-deleted variants, a set of isoform-specific PCR primers was used annealing at the junction of exons 0 and 3 (primer δ12) and the junction of exon 4 and 6 (primer δ5) or exon 4 and 7 (primer δ56), respectively, and identity of the resulting amplicons was confirmed by sequencing. Both cell lines exhibited a very weak expression of ERβ1, ERβδ125 and ERβδ1256 when compared to MDA-MB-231 breast cancer cells (Fig. 1c). Thus, we used these cell lines to elucidate the function of the exon-deleted ERβ splice variants in comparison to ERβ1 by means of overexpression. MCF-7 and SK-BR-3 cells were transfected with pTARGET mammalian expression vectors (Promega) containing the coding region of ERβ1, ERβ-δ125 or ERβ-δ1256 [34] or the original pTARGET vector as negative control. After verification of their expression in transient transfection assays on mRNA level by means of RT-PCR (data not shown), MCF-7 and SK-BR-3 clones stably expressing the transfected pTARGET derivatives were generated by G418 selection (300 μg/ml). About 6 weeks after transfection, 3 to 6 MCF-7 or SK-BR-3 clones per derivative were isolated using cloning disks and propagated. In these clones, mRNA levels of ERβ1, ERβδ125 or ERβδ1256, respectively, was relatively quantified in relation to β-actin expression by means of real time RT-PCR, avoiding false positive signals from vector DNA by comparison to a sample which was not reversely transcribed. The success of overexpression of these ERβ isoforms was additionally verified by sequencing of the amplified cDNA. MCF-7 and SK-BR-3 clones mock transfected with the original pTARGET vector as negative control were identified by detection of mRNA transcribed from the neomycin resistance gene of this vector by means of RT-PCR (primers pTAR1 and pTAR2). Additionally, overexpression of ERβ1, ERβδ125 or ERβδ1256 in the transfected clones was verified by means of immunoblot-analysis using ERβ antibody 5197P (Acris Antibodies, Hiddenhausen, Germany) (data not shown). Whereas we did not detect notable expression of all three protein isoforms in MCF-7 wildtype cells, MCF-7/ERβ1 cells exhibited a strong band of about 55 kDa, whereas the expected bands of 28 and 35 kDa size were detected in MCF-7 cells overexpressing ERβδ1256 or ERβδ125.
Three clones from each cell line stably exhibiting higher mRNA levels of the transfected ERβ subtypes similar the respective levels in MDA-MB-231 breast cancer cells (termed MCF-7 or SK-BR-3/β1H, MCF-7 or SK-BR-3/δ125H, MCF-7 or SK-BR-3/δ1256H) were chosen for further characterization. Since these three clones turned out to exhibit nearly identical properties in terms of proliferation, apoptosis, ERE activity and gene regulation, in the following data from the analysis of one clone is shown representatively.
Proliferation of breast cancer cells overexpressing ERβδ125, ERβδ1256 or ERβ1
Given that estrogen receptors are known to regulate cellular proliferation by different molecular mechanisms, we examined proliferation of our breast cancer models transfected with ERβ1 or the splice isoforms. For this purpose, both vector-transfected and ERβ-transfected clones from ERα-positive MCF-7 and estrogen-unresponsive SK-BR-3 cells were cultured in serumfree SR2 medium without E2 or in serumfree medium supplemented with 1 nM of this steroid hormone for up to five days. In the absence of E2, MCF-7 and SK-BR-3 cells overexpressing ERβ1 exhibited a significantly reduced proliferation when compared to growth of vector-transfected control cells, an effect which was more pronounced in the estrogen-unresponsive SK-BR-3 cell line (Fig. 2a, b). In contrast, overexpression of ERβδ125 οr ERβδ1256 did not affect cell growth of the breast cancer cell lines tested. Addition of 17β-estradiol as expected only increased growth of ERα-positive MCF-7, but not of SK-BR-3 control cells, and dose response analysis revealed that growth stimulation of MCF-7 cells was largest at a E2 concentration of 1 nM (Fig. 2c). This E2-triggered proliferation was observed in MCF-7 control cells and also in MCF-7 cells overexpressing the ERβδ125 or ERβδ1256 isoforms, but not in MCF-7 cells overexpressing ERβ1 (Fig. 2a). A similar growth stimulatory effect as triggered by E2 was observed after treatment of MCF-7 cells with selective ERα agonist PPT (1,3,5-tris (4-Hydroxyphenyl)-4-propyl-1H-pyrazole). Like E2, this substance was not able to stimulate proliferation of MCF-7 cells overexpressing ERβ1. In contrast, treatment with selective ERβ agonist DPN (diarylpropionitrile) clearly reduced cell growth of all MCF-7 clones tested. Whereas DPN slightly weakened proliferation of MCF-7 control cells and MCF-7 cells overexpressing the ERβδ125 or ERβδ1256 isoforms, cell numbers of ERβ1-overexpressing MCF-7 cells after 5 days of DPN treatment were even decreased down to 79% of the seeded cell number (Fig. 2a).

Growth of breast cancer cells overexpressing ERβδ125, ERβδ1256 or ERβ1. (a) Transfected MCF-7 cells overexpressing ERβ1, ERβδ125 or ERβδ1256 were cultured up to 5 days in serumfree (SR2) medium in the absence of estradiol or supplemented with 1 nM 17-β estradiol (E2), 100 nM of ERβ-agonist DPN or 100 nM of ERα-agonist PPT. Cell growth was compared to mock-transfected cells (MCF-7 con). Black circle = MCF-7-vector control; black triangle = MCF-7/ERβ1H; open rhombus = MCF-7/ERβδ1256H, open square = MCF-7/ERβδ125H. *P < 0.01 vs. vector transfected control cells. (b) Transfected SK-BR-3 cells overexpressing ERβ1, ERβδ125 or ERβδ1256 were cultured up to 5 days in serumfree medium in the absence of estradiol or supplemented with 1 nM 17-β estradiol (E2). Cell growth was compared to mock-transfected cells (SK-BR-3 con). Black circle = SK-BR-3-vector control; black triangle = SK-BR-3/ERβ1H; open rhombus = SK-BR-3/ERβδ1256H, open square = SK-BR-3/ERβδ125H. *P < 0.01 vs. vector transfected control cells. (c) Dose–response analysis of the generated MCF-7 and SK-BR-3 clones. Cells grown in serumfree (SR2) medium were treated with the indicated concentrations of 17β-estradiol and cell growth was determined on day 5. Generally, relative viable cell numbers were measured using the resazurin-based Cell Titer Blue fluorescence assay as described in the materials and methods section on day 0, 3, 4, and 5. Viable cell numbers are expressed as indicated in percentage of day 0 (a, b) or in percentage of the untreated control (c). Results were obtained from four separate experiments and are expressed as means ± SD
To examine, whether overexpression of the exon-deleted ERβ isoforms would be able to affect the response of breast cancer cell lines to selective estrogen receptor modulator tamoxifen, cells were treated with this substance (0.5–25 μM) in combination with 1 nM E2 for five days. Treatment with 4-OH tamoxifen clearly reduced cell numbers of all MCF-7 and SK-BR-3 clones tested in a dose-dependent manner, and response to this drug was much stronger in MCF-7 cells. However, overexpression of ERβ1 or the exon-deleted variants did not affect tamoxifen response of this cell line. In contrast, overexpression of ERβ1 in SK-BR-3 cells resulted in a slightly increased growth inhibitory effect of 4-OH tamoxifen (5 and 10 μM) (Fig. 3a).

Tamoxifen effects on breast cancer cells overexpressing ERβ1 or the exon-deleted splice variants. (a) For determination of antiproliferative action of tamoxifen, cells were incubated in serumfree medium supplemented with 1 nM E2 in combination with the indicated concentrations 4-OH tamoxifen for 5 days and relative cell growth was assessed by means of the Cell Titer Blue assay as described in the material and methods section. Growth is expressed in percentage of the E2-treated control (n = 3). (b) For examination of apoptotic effects of tamoxifen, cells cultured in serumfree medium supplemented with 1 nM E2 were treated with the indicated concentrations of 4-OH tamoxifen for 6 h and apoptosis was determined by measurement of caspase 3 or 7 activation by means of the luminometric Caspase-Glo 3/7 assay (Promega) according to the manufacturer′s protocol using a Victor3 multilabel counter (PerkinElmer, Germany). Apoptosis is expressed as means ± SD in percent of the untreated control cells (n = 3) *P < 0.05 vs. control cells
To examine, whether ERβδ125 or ERβδ1256 would be able to modulate function of ERβ1, we overexpressed these isoforms in a breast cancer cell line strongly expressing ERβ1. For this purpose, the transcript level of the exon-deleted variants in MDA-MB-231 cells was increased 20.9-fold (ERβδ125 or 34.7-fold (ERβδ1256) by means of transfection and G418 selection as described before. Proliferation assays with these MDA-MB-231 clones analyzing their growth in comparison to mock-transfected cells in the presence or absence of E2, DPN or 4-OH tamoxifen did not reveal any effect of overexpression of the exon-skipped isoforms (data not shown).
Apoptosis of breast cancer cells overexpressing ERβδ125, ERβδ1256 or ERβ1
Given that tamoxifen is known to trigger apoptosis in breast cancer cells and ERβ recently was reported to affect apoptosis in different cellular systems [37, 38], we also tested whether ERβ isoforms could modulate apoptotic response to tamoxifen. Cultured in serumfree medium supplemented with 1 nM E2, MCF-7 and SK-BR-3 cells overexpressing ERβ1, but not the exon-deleted isoforms exhibited an increased apoptosis rate when compared to control cells even in absence of tamoxifen. After treatment with different concentrations of this drug, a similarly elevated caspase activity of ERβ1- overexpressing cells was observed. In contrast, 4-OH tamoxifen concentrations from 0.5 to 10 μM did not trigger apoptosis in MCF-7 control cells or cells overexpressing the ERβδ125 or ERβδ1256 isoform, a significant apoptotic response of these MCF-7 and SK-BR-3 clones was only observed after treatment with 25 μM 4-OH tamoxifen (Fig. 3b).
Expression of estrogen-responsive genes in breast cancer cells overexpressing ERβδ125, ERβδ1256 or ERβ1
Given that estrogen receptors are ligand inducible transcription factors directly regulating gene transcription, we studied the effect of the exon-deleted ERβ splice variants in comparison to ERβ1 on expression of 14 estrogen responsive genes in MCF-7 and SK-BR-3 cells (progesterone receptor (PR), cyclin D1, cyclin A2, cyclin-dependent kinase 2 (CDK2), autotaxin, PS2, ERα, FAS ligand, HER2, cathepsin D, EGFR, IGFBP-4, WISP-2, p21 (WAF1). For this purpose, we analyzed expression of these genes in ERβ- or mock-transfected MCF-7 and SK-BR-3 cells cultured in serumfree medium or treated with 1 nM E2, 10 nM PPT or 10 nM DPN for 24 h on mRNA level by means of real time RT-PCR.
Four of the analyzed genes, p21(WAF1), cyclin A2, cyclin D1 and PR exhibited altered mRNA levels in ERβ1-transfected MCF-7 cells (Fig. 4). In ERα-positive MCF-7 cells, but not in SK-BR-3 cells, transcript levels of cell cycle regulator cyclin D1 were 2.4-fold induced after E2 treatment. Both in MCF-7 and SK-BR-3 cells, overexpression of ERβ1, but not of the exon-deleted isoforms resulted in a significantly reduced basal cyclin D1 mRNA level, which was not significantly increased after E2 treatment. Treatment with 5 μM 4-OH tamoxifen resulted in a decrease of cyclin D1 mRNA levels down to 40–50% in MCF-7 cells irrespective their ERβ status. Treatment with ERα-agonist PPT and ERβ-agonist DPN slightly increased cyclin D1 transcript levels in MCF-7 control cells and in cells overexpressing the exon-deleted variants, but decreased mRNA levels of this gene in MCF-7 cells overexpressing ERβ1.

Transcript levels of four genes in breast cancer cells overexpressing ERβδ125, ERβδ1256 or ERβ1. (a) SK-BR-3 clones cultured in serumfree medium were treated with 1 nM E2 24 h prior to total RNA isolation. (b) MCF-7 cells cultured in serumfree medium were treated with 100 nM DPN, 100 nM PPT or 1 nM E2 alone or in combination with 100 nM 4-OH tamoxifen for 24 h prior to total RNA isolation. Shown are the relative expression levels as determined by real time RT-PCR expressed in percentage of the corresponding β-actin transcript level (left panel) or as percentage of the solvent control (right panel). Results were obtained from five separate experiments and are expressed as means ± SD. *P < 0.05 vs. mock-transfected control cells
Cyclin A2 mRNA levels were 8.3-fold elevated after E2 treatment in MCF-7 cells, but not in SK-BR-3 cells. A similar E2-triggered increase of cyclin A2 transcript levels was observed in MCF-7 cells overexpressing the ERβδ125 or ERβδ1256 isoform, but overexpression of ERβ1 nearly abolished estrogenic cyclin A2 induction. In SK-BR-3 cells, overexpression of ERβ1, but not of the exon-deleted isoforms resulted in a significantly reduced basal cyclin A2 mRNA level, which was not significantly increased after E2 treatment. Treatment of MCF-7 clones with 4-OH tamoxifen did not affect cyclin A2 transcript levels in MCF-7 control and ERβδ1256-overexpressing cells, but reduced mRNA levels of this gene by about 50% in MCF-7 cells overexpressing ERβ1 or ERβδ125. Whereas treatment with DPN or PPT elevated cyclin A2 mRNA levels in vector-transfected MCF-7 cells and in MCF-7 cells overexpressing the exon-deleted isoforms, no such effect was observed in MCF-7 cells overexpressing ERβ1.
Transcript levels of antiproliferative cell cycle regulator and tumor suppressor p21(WAF1) were significantly increased in MCF-7 and SK-BR-3 cells overexpressing ERβ1, but E2 stimulation led to strong decrease of p21(WAF1) expression in MCF-7 cells. In contrast, overexpression of the ERβδ125 or ERβδ1256 isoform did not significantly affect p21 (WAF1) transcript levels which were not regulated by E2 in these clones and in MCF-7 control cells. Whereas tamoxifen treatment did not affect p21(WAF1) expression in all MCF-7 clones tested, treatment with ERβ agonist DPN resulted in a significant decrease of transcript levels, an effect which was even more pronounced in MCF-7/ERβ1 cells.
Progesterone receptor (PR) expression is known to be regulated by estrogens in an ERα-dependent manner. As expected E2 treatment resulted in a strong increase of PR mRNA levels in MCF-7 control cells. This effect was not significantly different in MCF-7 cells overexpressing the exon-deleted isoforms, but it was clearly diminished in MCF-7 cells overexpressing ERβ1. Tamoxifen treatment led to downregulation of PR mRNA levels by about 50% in MCF-7 control cells or cells expressing the ERβ splice variants, an effect which was even more pronounced in MCF-7 cells expressing ERβ1. Overexpression of ERβ1, but not of the exon-deleted isoforms diminished the PPT- or DPN-triggered upregulation of PR transcript levels.
Irrespective of treatment, we did not observe any significant differences between the different ERβ clones and MCF-7 or SK-BR-3 control cells regarding expression of CDK2, autotaxin, PS2, ERα, FAS ligand, HER2, cathepsin D, EGFR, IGFBP-4 or WISP-2.
Estrogen response element (ERE) activity in MCF-7 cells overexpressing ERβδ125, ERβδ1256 or ERβ1
To further confirm the observed lack of function of both exon-deleted isoforms, we analyzed their ability to modulate ERE activation by means of reporter gene assays. Like MCF-7 control cells, MCF-7 cells overexpressing ERβδ125 or ERβδ1256 exhibited a strong ERE activation as response to treatment with 1 nM E2 (Fig. 5). These cells also showed strong ERE activation after treatment with ERβ agonist DPN or ERα agonist PPT. In these cell lines, E2-triggered ERE activation was significantly inhibited by co-treatment with 0.5 μM 4-OH tamoxifen and to an even larger extent by co-treatment with pure antiestrogen ICI 182,780 (0.5 μM). In contrast, overexpression of ERβ1 totally inhibited ERE activation in MCF-7 cells.

Estrogen response element (ERE) activity in MCF-7 cells overexpressing ERβ1 or the exon-deleted variants. MCF-7 clones cultured in 6-well plates in serumfree (SR2) medium were transfected with 10 μg of reporter gene vector pTAL-ERE-SEAP (Clontech) and 5 μg of the pSV-β-GAL vector (Promega) as internal control for transfection efficacy. 24 h after transfection, cells were stimulated with the indicated substances. The next day, medium was removed and 20 μl of it were subjected to the Phospha Light Assay (Applied Biosystem) for luminometric quantification of secreted SEAP protein in the culture supernatant according to the instructions of the manufacturer. Cells were lysed using the Beta-Glo Assay (Promega) and subjected to this assay for luminometric determination of transfected β-galactosidase enzyme as internal control. Both luminometric SEAP and β-GAL quantification were carried out using a VICTOR3 multilabel plate reader (PerkinElmer). To normalize the data, SEAP values are expressed in relation to the measured β-GAL values (n = 5) *P < 0.01 vs. untreated control. *1 P < 0.01 vs. vector-transfected MCF-7 cells
Discussion
Recently we have identified the two novel human exon-deleted ERβ transcript variants ERβδ125 and ERβδ1256 in MDA-MB-231 breast cancer cells. Both transcripts previously have been demonstrated to be translated, they code for alternative ERβ proteins of about 35 kDa (ERβδ125) and 28 kDa (ERβδ1256) [39]. The aim of this study was to elucidate the function of both new ERβ isoforms in breast cancer cells with different ERα status in regulation of cell growth, apoptosis, gene expression and estrogen response element (ERE) activation. In contrast to MDA-MB-231 cells, both MCF-7 and SK-BR-3 breast cancer cells do not express notable amounts of ERβδ125 and ERβδ1256 transcripts. Whereas estrogen-responsive MCF-7 cells are a widely used model for hormone-dependent breast cancer expressing high levels of ERα and relatively low levels of ERβ, estrogen-unresponsive and HER-2 overexpressing SK-BR-3 cells do not express functional ERα and exhibit only marginal ERβ levels [40]. Our data demonstrating E2-triggered ERE activation in MCF-7, but not in SK-BR-3 cells confirm these differences on molecular level. However, the fact that treatment with ERβ agonist DPN was able to slightly reduce MCF-7 cell proliferation suggests that ERβ1 levels in these cell line are low but sufficient to mediate some weak effects of this ligand.
In this study, we stably introduced cDNA coding for ERβ1, ERβδ125 or ERβδ1256 into MCF-7 and SK-BR-3 cells to compare the effect of overexpression of these receptors in human breast cancer cells with different ERα status. For further characterization, we have chosen not the stably transfected clones exhibiting the highest expression levels, but the ones with lower overexpression levels comparable to the respective expression we measured in MDA-MB-231 cells and in single human breast cancer samples.
Several studies show evidence that ERβ negatively regulates cellular proliferation, promotes apoptosis and thus may have a protective role in normal breast and prostate [37, 41, 42]. Though also many ERβ splice variants are expressed both in mammary gland and in breast cancer [27], the specific role of ERβ splice isoforms in this tissue remains unclear. Three different ERβ variant mRNAs that have deletions in exon 5 or 6 or exons 5/6 have been identified in human breast, uterus and ovarian tissues [31, 43, 44]. A recent study examined the function of one of these exon-deleted variants, ERβ-δ5, suggesting that this isoform might act as a dominant negative receptor on ERα and ERβ pathways [38]. In another study, an ERβ isoform lacking the exons 2, 5 and 6 was identified and it was stated that deletion of these exons would cause a frame shift mutation resulting in premature termination of translation [45]. The exon-deleted variants ERβδ125 and ERβδ1256 we examined here use a different translation initiation codon in the beginning of exon 3 allowing translation in the same reading frame as ERβ1. The proteins coded by these variants are predicted not to contain the activation function 1 (AF-1) domain mediating the ligand-independent transcriptional activity of ERβ and are predicted to have deletions both in the DNA-binding domain (DBD) and in the ligand-binding domain (LBD). Thus, it is expected that both the ligand-dependent and ligand-independent activity of the deduced proteins are significantly diminished.
Our findings that ERβ1 promotes antitumoral effects on breast cancer cells in vitro are in line with previous studies reporting similar observations. Omoto et al. [46] stably expressed ERβ1 in MCF-7 cells under the control of a cytomegalovirus promotor and found that the receptor had a negative effect on proliferation of these cells and also reduced the number of colonies in an anchorage-independence assay. A similar study overexpressing ERβ1 in MCF-7 cells also reported growth inhibition by ERβ1 and reduced tumor formation in a mouse xenograft model [47]. ERβ1 overexpression was also shown to inhibit E2-triggered stimulation of estrogen-responsive T47D cells, an effect accompanied by decrease of Cyclin A2 and E mRNA levels, and also reduced angiogenesis and growth of T47D breast cancer xenografts [48, 49]. Other studies also suggested that antitumoral effects of ERβ are not necessarily dependent on the presence of ERα [37, 41]. Supporting these reports, in this study we demonstrate that ERβ1 exerts antitumoral effects not only on hormone-dependent MCF-7 breast cancer cells, but also on ERα-negative SK-BR-3 cells.
Recent studies suggest that many genes which are regulated by ERα are also regulated by ERβ in various tissues, but it is becoming increasingly clear that ERβ is also able to counteract ERα-triggered gene activation in some settings and also exerts specific gene regulation [50, 51]. A study examining the impact of ERβ on gene networks regulated by estrogen receptor alpha in breast cancer cells revealed that ERβ had diverse effects on gene expression, enhancing or counteracting ERα regulation for distinct subsets of estrogen target genes. Whereas ERβ in the absence of E2 elicited the stimulation or suppression of many genes that were normally only regulated by E2-triggered ERα activation, in the presence of this steroid ERβ elicited the expression of a unique group of genes that were not regulated by E2-triggered ERα activation [52]. Another recent study examining the effect of ERβ overexpression on transcriptome of ERα-positive T-47D breast cancer cells indentified a subset of 14 DNA replication and cell-cycle related genes to be down-regulated be ERβ [53]. In breast cancer cells, E2 is known to regulate expression of key cell cycle genes such as c-Myc, cyclin D1, cyclin E, cyclin A, cdc 25A, p45(Skip12) and p27(Kip1) [54–56]. The cyclin D promotor is one site where ERβ opposes ERα-mediated activations and the ERβ-antiestrogen complex can stimulate transcription [57]. Cyclin D transcription is known to be reduced by antiestrogens and its overexpression leads to resistance to antiestrogens [58, 59]. Cyclins E and A are important later in the G1 phase of the cell cycle when they participate in activation of CDK2, a crucial step in moving the cell into the S phase of cell cycle [60]. Cell cycle inhibitor and tumor suppressor gene p21 (WAF1) previously also has been demonstrated to be an important mediator of cellular estrogen response [61].
To analyse the molecular mechanisms underlying the observed alterations in proliferation and apoptosis of MCF-7 and SK-BR-3 cancer cells overexpressing ERβ1, we examined expression of a set of 14 estrogen-responsive genes on mRNA level. Overexpression of ERβ1 in MCF-7 cells previously was reported to inhibit proliferation by repressing c-myc, cyclin D1 and cyclin A expression and by induction of antiproliferative p21(WAF1) and p27(Kip1) leading to a G2 cell cycle arrest (Paruthiyil et al. 2004). Corresponding to these data, we observed both inhibition of E2-triggered upregulation of cyclin D1, cyclin A2 and PR in ERβ-overexpressing MCF-7 cells and ligand-independent decrease of cyclin D1 and increase of p21(WAF1) transcript levels. Our data also demonstrate ligand-independent inhibition of cyclin D1 and A2 expression and upregulation of p21(WAF1) in ERβ1-overexpressing SK-BR-3 breast cancer cells suggesting that ERβ1-triggered cell cycle arrest can also occur in an estrogen- and ERα-independent manner. Thus, gene regulation of cyclin D1, cyclin A2 and p21(WAF1) can be suggested to be important molecular mechanisms underlying the observed growth inhibitory effect of ERβ1 both in ERα-positive MCF-7 and in ERα-negative SK-BR-3 breast cancer cells. In contrast, overexpression of the exon-deleted ERβ variants did not significantly change mRNA levels of these cell cycle regulators, an observation explaining the lack of antiproliferative effects of these variants when overexpressed in breast cancer cells. PR gene transcript levels were not affected by ERβ1 overexpression in a ligand-independent manner, but ERβ1 blocked E2-triggered upregulation of this steroid hormone receptor, supporting the hypothesis that ERβ1 can modulate gene regulation both by ligand-dependent and ligand-independent mechanisms. In line with previous reports [58, 59], treatment of MCF-7, but not SK-BR-3 cells with tamoxifen resulted in inhibition of E2-triggered increase of cyclin D1 and PR mRNA levels. Tamoxifen effect on expression of these genes was not affected by overexpression of ERβ1 or the splice variants. In contrast, this drug did not affect p21(WAF1) mRNA levels and decreased cyclin A2 transcript levels only in MCF-7 cells overexpressing ERβ1 or the δ125 variant, an effect obviously too small to bear on growth of these cells. Whereas DPN or PPT did not affect gene expression in SK-BR-3 cells, both substances triggered increased transcript levels of cyclins A2 and D1 and PR and decreased p21 (WAF1) mRNA levels both in MCF-7 control cells and cells overexpressing one of the exon-deleted ERβ variants in a manner very similar to the observed E2 effect suggesting that the levels of ERα and ERβ1 present in these cells are sufficient to mediate agonistic effects on expression of these genes. The fact that DPN does not decrease cyclin D1 and A2 transcript levels in wildtype MCF-7 cells suggests that the inhibitory effect of ERβ1 on cyclin expression is dependent on higher ERβ1 expression levels. In contrast, treatment with these drugs did not increase mRNA levels of cyclins or PR in MCF-7 cells overexpressing ERβ1, also resembling the observed E2 effects. However, why overexpression of ERβ1 in breast cancer cells did not lead to stronger specific effects of DPN on gene expression in MCF-7 or SK-BR-3 cells remains unclear.
Further examining the molecular mechanisms underlying the observed differences between ERβ1 and the exon-deleted isoforms in regulation of gene expression, we analysed ERE activation in MCF-7 cells. Our data demonstrating a drastical inhibition of E2-, DPN- and PPT-triggered ERE activation by ERβ1 overexpression, but not by overexpression of ERβδ125 or ERβδ1256 confirms the inability of the exon-deleted isoforms to affect ERE activity. Our observation that DPN is able to trigger notable ERE activation in MCF-7 wildtype cells expressing low levels of ERβ1, whereas higher levels of ERβ1 do not mediate this DPN effect suggests that DPN effects on ERE activity in this cell line are strongly dependent on ERβ1 expression level.
Selective estrogen receptor modulator (SERM) tamoxifen is known to exert antitumoral effects on breast cancer cells both by inhibition of proliferation and by induction of cellular apoptosis. Whereas ERα is an established molecular marker for success of tamoxifen therapy, it remains controversial whether ERβ also is a predictor of endocrine therapy responsiveness in human breast cancer. Whereas tamoxifen binding to ERα is known to act antagonistically both at estrogen response elements (EREs) and AP-1 sites, binding of this drug to ERβ has agonistic effects on AP-1 sites. Our data showing no effect of ERβ1 or the novel isoforms on antiproliferative tamoxifen action in MCF-7 cells are in line with single clinical studies reporting no relevance of this receptor for endocrine responsiveness [25]. The observed slightly enhanced antiproliferative effect of tamoxifen on ERβ1-overepressing SK-BR-3 cells and the elevated apoptotic effect of this drug both on MCF-7 and SK-BR-3 cells overexpressing ERβ1, however, would support the majority of clinical studies reporting that patients with positive ERβ-status show better response to tamoxifen therapy [16, 20, 62]. Our results suggesting that ERβ1 has a stronger effect in potentiating tamoxifen-induced growth inhibition and apoptosis in ERα-negative SKBR-3 than in ERα-positive breast cancer cells also support a recent study showing a better response after adjuvant tamoxifen for ERβ-positive patients within the ERα-negative group than among ERα-positive patients [63]. If this holds true it would support the hypothesis that ERα-negative but ERβ-positive breast cancer patients could benefit from adjuvant tamoxifen.
In this study, we analyzed the function of two exon-deleted ERβ splice isoforms recently identified by our group in comparison to ERβ1 in breast cancer cell lines with different ERα status. Overexpression of ERβ1, but not of the exon-deleted variants exerted strong antitumoral effects both on ERα-positive MCF-7 and ERα-negative SK-BR-3 cells. Whereas ERβ1 overexpression slowed growth of MCF-7 and SK-BR-3 cells in the absence of E2 and also inhibited E2-triggered growth stimulation of MCF-7 cells, overexpression of the exon-skipped variants did not affect cell growth. Whereas overexpression of ERβ1 triggered an slightly increased antiproliferative effect of tamoxifen on SK-BR-3 cells and an increased basal apoptosis of MCF-7 and SK-BR-3 cells, an effect also present under tamoxifen treatment, the isoforms ERβδ125 or ERβδ1256 did not affect cellular tamoxifen response. The observed lack of function of the exon-deleted variants in terms of regulation of proliferation was accompanied both by their inability to affect expression of cyclins D1 and A2, p21 (WAF1) and PR and their disability to modulate ERE activation. In contrast, our results demonstrating antitumoral effects of ERβ1 on breast cancer cells with different ERα-status support the hypothesis that this receptor also is able to act as a tumor suppressor in an ERα-independent manner.
References
Ali S, Coombes RC (2002) Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2(2):101–112
Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-A (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 93(12):5925–5930
Leygue E, Dotzlaw H, Watson P, Murphy L (1998) Altered estrogen receptor alpha and beta mRNA expression during human breast tumorigenesis. Cancer Res 58:3197–3201
Jarvinen T, Pelto-Huikko M, Holli K, Isola J (2000) Estrogen receptor beta is coexpressed with ERalpha and PR and associated with nodal status grade and proliferation rate in breast cancer. Am J Pathol 156:29–35
Forster C, Makela S, Warri A, Kietz S, Becker D, Hultenby K, Warner M, Gustafsson J (2002) Involvement of estrogen receptor b in terminal differentiation of mammary gland epithelium. PNAS 99:15578–15583
Hall J, McDonnell D (1999) The estrogen receptor betaisoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566–5578
Weihua Z, Saji S, Makinen S, Cheng G, Jensen E, Warner M, Gustaffson JA (2000) Estrogen receptor (ER) b a modulator of ERa in the uterus. PNAS 97:5936–5941
Peng B, Lu B, Leygue E, Murphy L (2003) Putative functional characteristics of human estrogen receptor-beta isoforms. J Mol Endocrinol 30:13–29
Roger P, Sahla M, Makela S, Gustafsson J.A, Baldet P, Rochefort H (2001) Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res 61:2537–2541
Skliris G, Munot K, Bell S, Carder P, Lane S, Horgan K, Lansdown M, Parkes A, Hanby A, Markham A, Speirs V (2003) Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyltransferase inhibitors in a cell line model. J Pathol 201:213–220
Dotzlaw H, Leygue E, Watson P, Murphy L (1999) Estrogen receptor-b messenger RNA expression in human breast tumor biopsies: relationship to steroid receptor status and regulation by progestins. Cancer Res 59:529–532
Speirs V (2002) Oestrogen receptor beta in breast cancer: good bad or still too early to tell? J Pathol 197:143–147
Fuqua S, Schiff R, Parra I, Friedrichs W, Su J, McKee D, Slentz-Kesler K, Moore L, Willson T, Moore J (1999) Expression of wild-type estrogen receptor beta and variant isoforms in human breast cancer. Cancer Res 59:5425–5428
Saji S, Omoto Y, Shimizu C, Warner M, Hayashi Y, Horiguchi S, Watanabe T, Hayashi S, Gustafsson J (2002b) Expression of estrogen receptor (ER) (beta)cx protein in ER(alpha)-positive breast cancer: specific correlation with progesterone receptor. Cancer Res 62:4849–4853
Saji S, Omoto Y, Shimizu C, Horiguchi S, Watanabe T, Funata N, Hayash S, Gustafsson J, Toi M (2002a) A clinical impact of assay of estrogen receptor beta cx in breast cancer. Breast Cancer Res Treat 9:303–307
Mann S, Laucirica R, Carlson N, Younes PS, Ali N, Younes A, Li Y, Younes M (2001) Estrogen receptor beta expression in invasive breast cancer. Hum Pathol 32(1):113–118
Murphy L, Cherlet T, Lewis A, Banu Y, Watson P (2003) New insights into estogen receptor function in human breast cancer. Ann Med 35:614–631
Esslimani-Sahla M, Simony-Lafontaine J, Kramar A, Lavaill R, Mollevi C, Warner M, Gustafsson J-A, Rochefort H (2004) Estrogen receptor beta (ERbeta) level but not its ERbeta-cx variant helps to predict tamoxifen resistance in breast cancer. Clin Cancer Res 10:5769–5776
Fleming F, Hill A, McDermott E, O’Higgins N, Young L (2004) Differential recruitment of coregulator proteins steroid receptor coactivator-1 and silencing mediator for retinoid and thyroid receptors to the estrogen receptorestrogen response element by beta-estradiol and 4-hydroxytamoxifen in human breast cancer. J Clin Endocrinol Metab 89:375–383
Hopp TA, Weiss HL, Parra IS, Cui Y, Osborne CK, Fuqua SA (2004) Low levels of estrogen receptor beta protein predict resistance to tamoxifen therapy in breast cancer. Clin Cancer Res 10(22):7490–7499
Saji S, Hirose M, Toi M (2005) Clinical significance of estrogen receptor beta in breast cancer. Cancer Chemother Pharmacol 56:21–26
Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Khosla S, Spelsberg TC (2005) Estrogen receptor alpha and beta heterodimers exert unique effects on estrogen- and tamoxifen-dependent gene expression in human U2OS osteosarcoma cells. Mol Endocrinol 19(6):1555–1568
Osborne C (1998) Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat 51:227–238
Jensen E, Cheng G, Palmieri C, Saji S, Makela S, Van Noorden S, Wahlstrom T, Warner M, Coombes R, Gustafsson J-A (2001) Estrogen receptors and proliferation markers in primary and recurrent breast cancer. PNAS 98:15197–15202
O’Neill P, Davies M, Shaaban A, Innes H, Torevell A, Sibson D, Foster C (2004) Wild-type oestrogen receptor beta (ERbeta1) mRNA and protein expression in Tamoxifen treated post-menopausal breast cancers. Br J Cancer 91:1694–1702
Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, Muramatsu M (1998) Molecular cloning and characterization of human estrogen receptor bcx: potential inhibitor of estrogen action in human. Nucleic Acids Res 26:3505–3512
Poola I, Abraham J, Liu A (2002a) Estrogen receptor beta splice variant mRNAs are differentially altered during breast carcinogenesis. J Steroid Biochem Mol Biol 82(2–3):169–179
Price RH Jr, Butler CA, Webb P, Uht R, Kushner P, Handa RJ (2001) A splice variant of estrogen receptor beta missing exon 3 displays altered subnuclear localization and capacity for transcriptional activation. Endocrinology 142(5):2039–2049
Herynk MH, Fuqua SA (2004) Estrogen receptor mutations in human disease. Endocr Rev 25(6):869–898
Price RH Jr, Lorenzon N, Handa RJ (2000) Differential expression of estrogen receptor beta splice variants in rat brain: identification and characterization of a novel variant missing exon 4. Mol Brain Res 80(2):260–268
Speirs V, Adams IP, Walton DS, Atkin SL (2000) Identification of wild-type and exon 5 deletion variants of estrogen receptor beta in normal human mammary gland. J Clin Endocrinol Metab 85(4):1601–1605
Zhao C, Toresson G, Xu L, Koehler KF, Gustafsson JA, Dahlman-Wright K (2005) Mouse estrogen receptor beta isoforms exhibit differences in ligand selectivity and coactivator recruitment. Biochemistry 44(22):7936–7944
Sierens JE, Scobie GA, Wilson J, Saunders PT (2004) Cloning of oestrogen receptor beta from Old and New World primates: identification of splice variants and functional analysis. J Mol Endocrinol 32(3):703–718
Treeck O, Pfeiler G, Horn F, Federhofer B, Houlihan H, Vollmer A, Ortmann O (2007) Novel estrogen receptor beta transcript variants identified in human breast cancer cells affect cell growth and apoptosis of COS-1 cells. Mol Cell Endocrinol 264:50–60
Ståhlberg A, Åman P, Ridell B, Mostad P, Kubista M (2003) A quantitative real-time PCR method for detection of B-Lymphocyte monoclonality by comparison of kappa and lambda Immunoglobulin Light Chain Expression. Clin Chem 49(1):51–59
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2^( -delta delta Ct ) method. Methods 25(4):402–408
Cheng J, Lee EJ, Madison LD, Lazennec G (2004) Expression of estrogen receptor beta in prostate carcinoma cells inhibits invasion and proliferation and triggers apoptosis. FEBS Lett 566(1–3):169–172
Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA (2005) Estrogen receptors alpha (ERalpha) and beta (ERbeta) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 24(44):6605–6616
Treeck O, Pfeiler G, Mitter D, Lattrich C, Piendl G, Ortmann O (2007) Estrogen receptor β1 exerts antitumoral effects on SK-OV-3 ovarian cancer cells. J Endocrinol 193(3):421–433
Vladusic EA, Hornby AE, Guerra-Vladusic FK, Lakins J, Lupu R (2000) Expression and regulation of estrogen receptor beta in human breast tumors and cell lines. Oncol Rep 7(1):157–167
Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F (2001) ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology 142(9):4120–4130
Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A (2005) The androgen derivative 5alpha-androstane-3beta, 17beta-diol inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype. Cancer Res 65(12):5445–5453
Lu B, Leygue E, Dotzlaw H, Murphy LJ, Murphy LC, Watson PH (1998) Estrogen receptor-beta mRNA variants in human and murine tissues. Mol Cell Endocrinol 138(1–2):199–203
Vladusic EA, Hornby AE, Guerra-Vladusic FK, Lupu R (1998) Expression of estrogen receptor beta messenger RNA variant in breast cancer. Cancer Res 58(2):210–214
Poola I, Abraham J, Baldwin K (2002b) Identification of ten exon deleted ERbeta mRNAs in human ovary, breast, uterus and bone tissues: alternate splicing pattern of estrogen receptor beta mRNA is distinct from that of estrogen receptor alpha. FEBS Lett 516(1–3):133–138
Omoto Y, Eguchi H, Yamamoto-Yamaguchi Y, Hayashi S (2003) Estrogen receptor (ER) beta1 and ERbetacx/beta2 inhibit ERalpha function differently in breast cancer cell line MCF7. Oncogene 22:5011–5020
Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC (2004) Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res 64(1):423–428
Hartman J, Lindberg K, Morani A, Inzunza J, Strom A, Gustafsson JA (2006) Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res 66(23):11207–11213
Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA (2004) Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA 101(6):1566–1571
Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C (2003) Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription,supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol 17(2):203–208
Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS (2004) Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) alpha or ERbeta in human osteosarcoma cells: distinct and common target genes for these receptors. Endocrinology 145(7):3473–3486
Chang EC, Frasor J, Komm B, Katzenellenbogen BS (2006) Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 147(10):4831–4842
Lin CY, Ström A, Li Kong S, Kietz S, Thomsen JS, Tee JB, Vega VB, Miller LD, Smeds J, Bergh J, Gustafsson JA, Liu ET, (2007) Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Res 9(2):R25
Carroll JS, Swarbrick A, Musgrove EA, Sutherland RL (2002) Mechanisms of growth arrest by c-myc antisense oligonucleotides in MCF-7 breast cancer cells: implications for the antiproliferative effects of antiestrogens. Cancer Res 62:3126–3131
Watts CK, Sweeney KJ, Warlters A, Musgrove EA, Sutherland RL (1994) Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res Treat 31:95–105
Foster JS, Henley DC, Bukovsky A, Seth P, Wimalasena J (2001) Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function. Mol Cell Biol 21:794–810
Liu MM, Albanese C, Anderson CM, Hilty K, Webb P, Uht RM, Price RH Jr, Pestell RG, Kushner PJ (2002) Opposing action of estrogen receptors alpha and beta on cyclin D1 gene expression. J Biol Chem 277(27):24353–24360
Hodges LC, Cook JD, Lobenhofer EK, Li L, Bennett L, Bushel PR, Aldaz CM, Afshari CA, Walker CL (2003) Tamoxifen functions as a molecular agonist inducing cell cycle-associated genes in breast cancer cells. Mol Cancer Res 1:300–311
Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL (2002) Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res 62:6916–6923
Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL (2003) Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer 10:179–186
Thomas TJ, Faaland CA, Adhikarakunnathu S, Watkins LF, Thomas T (1998) Induction of p21 (CIP1/WAF1/SID1) by estradiol in a breast epithelial cell line transfected with the recombinant estrogen receptor gene: a possible mechanism for a negative regulatory role of estradiol. Breast Cancer Res Treat 47(2):181–193
Iwase H, Zhang Z, Omoto Y, Sugiura H, Yamashita H, Toyama T, Iwata H, Kobayashi S (2003) Clinical significance of the expression of estrogen receptors alpha and beta for endocrine therapy of breast cancer. Cancer Chemother Pharmacol 52(Suppl 1):34–38
Gruvberger-Saal SK, Bendahl PO, Saal LH, Laakso M, Hegardt C, Eden P, Peterson C, Malmström P, Isola J, Borg A, Fernö M (2007) Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin Cancer Res 13(7):1987–1994
Acknowledgements
We thank Angelika Vollmer, Gerhard Piendl and Helena Houlihan for expert technical assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material
Rights and permissions
About this article
Cite this article
Treeck, O., Juhasz-Boess, I., Lattrich, C. et al. Effects of exon-deleted estrogen receptor β transcript variants on growth, apoptosis and gene expression of human breast cancer cell lines. Breast Cancer Res Treat 110, 507–520 (2008). https://doi.org/10.1007/s10549-007-9749-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-007-9749-7