
Abstract
Introduction
Alpha-1,3-glucosyltransferase congenital disorder of glycosylation (ALG6-CDG) is a congenital disorder of glycosylation. The original patients were described with hypotonia, developmental disability, epilepsy, and increased bleeding tendency.
Methods
Based on Euroglycan database registration, we approached referring clinicians and collected comprehensive data on 41 patients.
Results
We found hypotonia and developmental delay in all ALG6-CDG patients and epilepsy, ataxia, proximal muscle weakness, and, in the majority of cases, failure to thrive. Nine patients developed intractable seizures. Coagulation anomalies were present in <50 % of cases, without spontaneous bleedings. Facial dysmorphism was rare, but seven patients showed missing phalanges and brachydactyly. Cyclic behavioral change, with autistic features and depressive episodes, was one of the most significant complaints. Eleven children died before the age of 4 years due to protein losing enteropathy (PLE), sepsis, or seizures. The oldest patient was a 40 year-old Dutch woman. The most common pathogenic protein alterations were p.A333V and p.I299Del, without any clear genotype–phenotype correlation.
Discussion
ALG6-CDG has been now described in 89 patients, making it the second most common type of CDG. It has a recognizable phenotype and a primary neurologic presentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital Disorders of Glycosylation (CDG) is a group of inborn errors of metabolism with ~80 known biochemical subtypes of secretory glycosylation defects. These anomalies are easily diagnosed by plasma or serum glycan analysis (Lefeber et al. 2011; Scott et al. 2014). After phosphomannomutase 2 (PMM2)-CDG, the second most common CDG is alpha-1,3-Glucosyltransferase (ALG6)-CDG (previously called CDG Ic), a defect in N-glycan assembly in the endoplasmic reticulum (ER). ALG6-CDG is caused by an autosomal recessive defect of the enzyme Man9GlcNAc2-P-P-Dol α-1,3-glucosyltransferase. A genetic defect in ALG6 leads to an accumulation of Man9GlcNAc2-P-P-Dol and other lipid-linked oligosaccharides without glucose residues and subsequent hypoglycosylation of serum glycoproteins (Jaeken et al. 2015; Lefeber et al. 2011). Serum transferrin isoelectric focusing (TIEF), or/and mass-spectrometric analysis of transferrin glycoforms, is the first step in the screening analysis. Further analysis of lipid-linked oligosaccharides is possible in fibroblasts. Direct genetic analysis confirms the diagnosis (Lefeber et al. 2011).
The ALG6 gene is highly conserved throughout evolution, from mammals to yeast. In humans, it is localized on chromosome 1p31.3 (MIM 603147 and MIM 604566). Missense mutations p.A333V and p.I299Del are recognized as common mutations in ALG6-CDG. Patients with these mutations were described with a phenotype of hypotonia, speech disability, and epilepsy (Lefeber et al. 2011). So far, its clinical course and progression is reported in 48 ALG6-CDG patients showing severe psychomotor retardation, seizures, speech disability, some dysmorphic symptoms, and variable presence of failure to thrive, gastrointestinal features, hormonal anomalies, coagulopathy, and elevated transaminase activities (Al-Owain et al. 2010; Dercksen et al. 2013; Drijvers et al. 2010; Ichikawa et al. 2013; Lefeber et al. 2011; Miller et al. 2011). Unique features, such as abnormal skeletal development and short fingers, have been described in two ALG6-CDG patients, suggesting a possible association between skeletal development and the primary glycosylation defect (Drijvers et al. 2010). In this paper, we review the clinical presentation, neurologic features, unique features, and genetic background of 41 patients diagnosed with ALG6-CDG.
Methods
We retrospectively evaluated the clinical data of 41 patients diagnosed with ALG6-CDG between 1995 and 2013 either registered by the Euroglycan database or followed by a clinician involved in Euroglycanet and participating in the reported study. Patients originated from The Netherlands, Germany, Ireland, Belgium, France, Arabia, Turkey, Montenegro, and South Africa (Caucasian). Patients with mixed ethnic backgrounds originated from Brazil, Portugal, North Africa and Italy. Metabolic physicians caring for these patients were approached through the Euroglycan patient registration database (University of Leuven) and the national metabolic patient registry in The Netherlands.
Diagnostic criteria included a biochemically confirmed N-glycosylation disorder with lipid-linked oligosaccharide precursor (LLO) results typical for an ALG6 defect and/or confirmed by mutation analysis. From this cohort, 35 patients have never been reported, and six were recently reported but the authors offered follow-up data (Al-Owain et al. 2010; Dercksen et al. 2013). Six patients with insufficient data (no availability of results of mutation analysis but with type I TIEF profile and characteristic LLO abnormalities for ALG6) were excluded from the genotype–phenotype analysis. Clinical data of these patients were included for clinical data evaluation and long-term prognosis assessment, even when mutation results were not available.
Physicians of participating patients were interviewed by a standard questionnaire. Patients’ medical history, genetic information, and all clinical features, which have been previously observed in different forms of CDG-I, were included in our survey. Further information was requested upon receiving information on unusual features. Certain questions were adapted for specific age groups (developmental assessment, intellectual and speech assessments, Child-Behavior Checklist, and quality of life assessment). Genetic data were collected from patients in the Euroglycan database and from participating physicians, including additional data obtained by the survey. Genetic data were evaluated in 35 patients enrolled in the clinical study. Due to the presence of common mutations in ALG6-CDG, the genotype–phenotype correlation was analyzed by dividing the patients into three groups: p.A333V homozygous patients, p.A333V compound heterozygous patients, and patients homozygous for a severe mutation. A mutation was labeled a priori as severe according to previously reported severe or deleterious phenotypic expression and according to type and localization of the mutation in the ALG6 gene (Dercksen et al. 2013).
We additionally evaluated the roles of the p.F304S mutation, which has been hypothesized to be a variant in ALG6, affecting PMM2 gene expression, PMM2-CDG phenotype severity (Westphal et al. 2000), and the role of the known ALG6 gene polymorphism p.Y131H. This variant is hypothesized to influence glycosylation (Supplementary Table 1).
Results
Clinical features
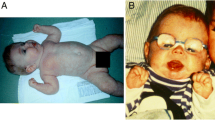
We obtained clinical data from 41 ALG6-CDG patients, including information on psychomotor development, growth, neurological problems, other organ involvement, systemic manifestations, malformations, and dysmorphic features or additional symptoms (Figs. 1, Tables 1, 2, 3 and 4). Six of these 41 patients had no mutation data available. Most patients (32) were of Caucasian background. At the time of inquiry, mean patient age was 11 years and 8 months. Two patients were first cousins; otherwise, patients were unrelated. (There is a possible genetic founder effect in the South African group). Eleven patients died in the first 4 years of life due to sepsis, protein-losing enteropathy (PLE), or intractable epilepsy. We found abnormal development/disability (n = 41) and hypotonia (n = 40) as the most common symptoms. Hypotonia was always axial and was associated with proximal muscle weakness. In most cases, generalized hypotonia was observed during the first 4–6 years of age.

Distribution of clinical features in the patient cohort of 42 alpha-1,3-glucosyltransferase congenital disorders of glycosylation (ALG6-CDG) cases
Epilepsy was present in the majority of our patients (n = 30). All patients had infantile presentations presenting between the ages of 3 and 11 months. At onset, most children had myoclonic seizures or infantile spasms. EEG showed slowed background rhythm, and generalized epileptic activity in most cases, except for one patient, who was almost 1 year old at presentation and had focal seizures. The seizure pattern changed with age, and one fourth of patients developed therapy-resistant epilepsy. There was no clear developmental regression in patients developing tonic/clonic or myoclonic seizures. Intractable seizures, mostly West syndrome, and epileptic encephalopathy were described in nine of 41 patients. Of those nine patients, two were trialed on adrenocorticotropic hormone (ACTH) therapy, one had a ketogenic diet without success, and the other patients were on vigabatrin, valproic acid, and lamotrigine therapy. Electroencephalogram (EEG) showed mostly multifocal epileptic activity, burst suppression pattern in four cases, and status epilepticus in three. The nine patients with intractable seizures all had severe developmental delay and profound speech delay, most developed microcephaly, and there was a high lethality rate.
Additionally we found ataxia (n = 20), nystagmus (n = 18) ,and strabismus (n = 25) in about half of the patients. Only six of the 20 patents with ataxia showed vermis hypoplasia and/or cerebellar atrophy on magnetic resonance (MR) brain imaging. Other MRI findings included brain atrophy in several patients, corpus callosum hypoplasia in one case, and Dandy Walker malformation with vermis hypoplasia in another one.
All patients had some degree of developmental delay, and speech disability was present in all but one patient older than 3 years of age (n = 34). Behavioral problems or mood disorders occurred in 14 patients, including autistic features, autism, periodic mood changes, episodes of aggressive behavior, and sleep disturbance. Although most of these 14 patients showed autistic features, including abnormal communication, difficulties with social interaction, and recurrent episodes of repetitive behavior, only five patients were diagnosed with autism that showed classic diagnostic features and regression in speech and development between the age of 3 and 4 years. Aggressive behavior, mostly around puberty, was present in five cases. Sleep disturbance requiring daily melatonin treatment was noted in seven. In 25 children, failure to thrive necessitated an increased caloric intake (special diet). Six patients had PLE. Seven patients also had anemia, most likely due to malabsorption. Four PLE patients followed an elementary diet due PLE, and two received octreotide injections to decrease enteral protein loss. Skeletal abnormalities present in 15 children were brachydactyly, (Figure 2) arachnodactyly, limited extension of joints, short arms, and scoliosis. Three patients had dilated cardiomyopathy, and one patient hepatosplenomegaly. Dysmorphic features in 15 patients were unspecific (deep-set eyes, bitemporal narrowing, broad nasal bridge, short philtrum, and hypertelorism in individual cases). Rare clinical symptoms were a short umbilical cord (n = 2), ptosis (n = 1), and abnormal fat distribution (n = 4). Laboratory abnormalities were not systematically assessed, and most children had only screening laboratory data for thyroid hormone levels, transaminase levels, blood glucose, and bleeding time in early stages of the disease, which was repeated only if an abnormal level was found initially. Patients with PLE had a more extensive workup, showing severe hypoalbuminemia, significantly elevated serum transaminases, low immunoglobulin G (IgG) levels, and anemia. Two patients had recurrent hypoglycemia. Hyperinsulinism was found in two other patients with episodes of hypoglycemia. One patient had additionally decreased cortisol and ACTH levels. Bleeding (increased bleeding time and easy bruising) was less common than expected (n = 18) and were mainly subclinical. Only one patient experienced thrombotic episodes; no comprehensive laboratory data were available on hemostasis. Abnormal coagulation was demonstrated mostly by a prolonged bleeding time. Twelve patients underwent minor surgery (strabismus operation or tooth extraction) without complications. Three of the 12 had decreased factor IX and antithrombin III levels.

Distal limb malformation in a patient with Brachydactyly of the 2-5th toes
Genetic properties
From the 60 ALG6-CDG patients registered before 2013 by the Euroglycan network for whom mutation data were available, we found four thus far unreported mutations in addition to the 23 different ALG6 mutations described previously (www.lovd.nl/ALG6). The four new mutations were missense mutations located in exon 3 (p.H79R and p.A84T), exon 5 (p.D120G), and exon 10 (p.S308N). The pathogenic change leading to p.A333V was the most common, with 26 alleles in 15 patients, followed by p.I299Del (12 alleles in eight patients) and c.257 + 5G > A (seven alleles in seven patients). Homozygosity was seen with mutations related to p.A333V, p.A84T, p.I299Del, and p.Y161C. Mutations c.257 + 5G > A and protein changes p.Y57X, p.G227E, p.V447SFsX44, and R18Q, were only found in compound heterozygous patients. Alterations p.R18Q and p.Y57X were found once. In all patients, mutation c.257 + 5G > A was found seven times as compound heterozygous, while p.G227E was detected five times in a compound heterozygous form.
Genotype–phenotype analysis
For the genotype–phenotype analysis, we analyzed 22 patients who were divided into three groups based on their genotype. Group 1 comprised 11 patients homozygous for the change p.A333V. Group 2 comprised four p.A333V compound heterozygous patients, and group 3 comprised seven patients homozygous for any mutation other than p.A333V (patients 13, 15, 19, 21, 28, 33, 34). All patients were included for clinical analysis according to the previously described criteria. In the first group, three of the 11 patients died; in the second group, two of four; and in the third group, one.
Neurological symptoms and developmental disability
Speech, developmental or intellectual disability, muscle involvement, and hypotonia were present in all patients. Muscle weakness, infantile seizures, and wheelchair dependence was significant (≥50 % present) in all three groups. In the group homozygous for p.A333V, all patients had severe delay in speech development. Even patients with mild motor delay had mostly severe speech delay or absent speech. One patient homozygous for p.I299del and another heterozygous for p.I299del had relatively mild speech delay. Brain malformations were found mostly in the second group (50 %) and ataxia mostly in groups 1 (91 %) and 3 (50 %). Visual loss was highly represented in the p.A333V compound heterozygous group: 75 % compared with 9 % (group 1) and 30 % (group 3). Strabismus was less frequent in group 2 (25 %), and nystagmus was unique in group 3. Microcephaly was found most frequently in the p.A333V homozygous group: 55 % compared with groups 2 and 3 at 0 % and 1 %, respectively.
Physical appearance
There was no correlation between a specific mutation and dysmorphic features. Failure to thrive was common in groups 1 and 2 (82 % and 100 %, respectively). Being underweight and of short stature were more prevalent in group 1 (82 %) than in groups 3 (30 %) and 2 (0 % and 25 % respectively).
Systemic involvement and laboratory abnormalities
PLE was found in 75 % of compound heterozygous patients and only in 9 % of p.A333V homozygous patients, and in no patient homozygous for a mutation other than leading to change p.A333V. Increased bleeding tendency was one of the most common features, with 73 % in group 1 and 50 % in group 3. Only one of four patients with compound heterozygosity had bleeding anomaly. Hypoglycemia and increased serum transaminases were also most prevalent in p.A333V homozygous patients. In summary, the most severe phenotype was observed in p.A333V homozygous patients and comprised hypotonia, intellectual disability, severe proximal muscle weakness, seizures, visual or hearing loss, failure to thrive, and PLE; however, the relatively small group 2 had the highest mortality rate. The patient with a severe neurological phenotype carrying the frameshift mutation p.V447SfsX44 died within the first 5 months of life due to intractable PLE. We found the additional presence of the p.F304S variant in one patient in our cohort, without any specific correlation with ALG6-CDG phenotype severity. Although the pathogeneicity of the p.F304S variant has been debated before, due to its very high frequency and low conservation, it has been defined as a polymorphism (Vuillaumier-Barrot et al. 2001; Westphal et al. 2003). The only patient carrying this alteration additional to the p.I299del homozygous state has a phenotype that is comparable with other p.I299del-homozygous patients. The additional presence of p.Y131H, which is a known polymorphism, showed no correlation with the phenotype in our patients with ALG6-CDG. The variant was found in three patients with different genotypes, and no correlation was detected with phenotype severity (Goreta et al. 2012) (Supplementary Table 1).
Discussion
In this paper, we redefine the phenotype of ALG6-CDG in 41 patients. In this patient cohort, the most common pathogenic changes were p.A333V and p.I299Del. Patients homozygous for p.A333V seemed to have the most severe presentation and the most multisystem involvement, but the subgroups were small, and we found no clear genotype–phenotype correlation. Phenotypic variability was present between siblings as well.
Epilepsy is reported in >70 % of patients, and in our cohort, it presented as one of the most frequent neurological features, appearing in infancy and intractable in one fourth of patients. Besides the previously reported triad of hypotonia, developmental disability, and seizures, we also found a high percentage of other neurological symptoms, including absent speech, ataxia, proximal muscle weakness, and visual loss. Several patients had behavioral abnormalities, including autistic behavior, mood swings, and periods of inertia alternating with periods of aggressive behavior, which is unusual for most CDG-I patients. Special features included facial dysmorphism, cardiomyopathy, and limb malformations in several patients. Some patients had severe distal limb reduction. Abnormal fat distribution was present in few patients. Laboratory abnormalities were present in about half of the patients, although not all patients were screened systematically. Anemia was common as well. The most common alteration was an increased bleeding time, although less than half of the ALG6-CDG patients showed an increased bleeding tendency. In some early-onset cases with severe multisystem involvement, PLE, hypoalbuminemia, hypoglycemia, and frequent infections were present and led to early lethality. Interestingly, no fetal edema or fetal hydrops was observed in this cohort.
Some patients showed stagnation in psychomotor development around puberty, but there was no clear regression. Most patients are still young, which makes long-term prognosis difficult. A few older patients (12–16 years) developed significant behavioral abnormalities in this period, and some of them developed hypothyroidism or had new-onset of episodes of hypoglycemia. According to our data, most ALG6-CDG patients reached puberty and a few reached adulthood. The oldest patient was a 40-year-old Dutch woman. She was able to make some steps independently, had mild speech disability, and had static encephalopathy. PLE, one of the most significant life-threatening symptom of ALG6-CDG, was successfully treated with a low-fat/elementary protein diet and subcutaneous treatment with octreotide. Anemia was a frequent comorbidity. Chronic intestinal protein loss and hypogammaglobulinemia might lead to early lethality in young ALG6-CDG patients. We found a high percentage of associated laboratory abnormalities in our patients, necessitating prospective, regular screening of glycemia, coagulation, and endocrine and liver function.
We detected the mutations p.A333V and p.I299del in half of our patients, Allele frequencies for the most common mutations in our patient group, according to the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/), is at 0.00006595 for p.A333V and 0.000008268 for p.I299del. Surprisingly, for c.257 + 5G > A, database frequency was as high as 0.0004560. While the relative frequency of p.A333V and p.I299del were somewhat comparable to the database’s allele frequency, we encountered in proportion significantly less patients with the mutation c.257 + 5G > A compared with that previously reported.
Including patients in our cohort, there are 89 ALG-CDG patients reported in the literature (Jaeken et al. 2015), and the ALG6 defect remains the second most common CDG type, with a recognizable phenotype. Since some children only present with hypotonia and seizures, or behavioral abnormality and speech disability, we recommend systematic glycosylation screening in developmental disability.
Abbreviations
- ALG6:
-
Alpha-1,3-glucosyltransferase
- CDG:
-
Congenital disorder of glycosylation
- TIEF:
-
Transferrin isoelectric focusing
- PLE:
-
Protein-losing enteropathy
References
Al-Owain M, Mohamed S, Kaya N, Zagal A, Matthijs G, Jaeken J (2010) A novel mutation and first report of dilated cardiomyopathy in ALG6-CDG (CDG-Ic): a case report. Orphanet J Rare Dis 5:7
Dercksen M, Crutchley AC, Honey EM, Lippert MM, Matthijs G, Mienie LJ, Schuman HC, Vorster BC, Jaeken J (2013) ALG6-CDG in South Africa: genotype-phenotype description of five novel patients. JIMD Rep 8:17–23
Drijvers JM, Lefeber DJ, de Munnik SA, Pfundt R, van de Leeuw N, Marcelis C, Thiel C, Koerner C, Wevers RA, Morava E (2010) Skeletal dysplasia with brachytelephalangy in a patient with a congenital disorder of glycosylation due to ALG6 gene mutations. Clin Genet 77:507–509
Goreta SS, Dabelic S, Pavlinic D, Lauc G, Dumic J (2012) Frequency determination of α-1,3 glucosyltransferase p.Y131H and p.F304S polymorphisms in the croatian population revealed five novel single nucleotide polymorphisms in the hALG6 gene. Genet Test Mol Biomark 16:50–53
Ichikawa K, Kadoya M, Wada Y, Okamoto N (2013) Congenital disorder of glycosylation type Ic: report of a Japanese case. Brain Dev 35:586–589
Jaeken J, Lefeber D, Matthijs G (2015) Clinical utility gene card for: ALG6 defective congenital disorder of glycosylation. Eur J Hum Genet 23. doi: 10.1038/ejhg.2014.146
Lefeber DJ, Morava E, Jaeken J (2011) How to find and diagnose a CDG due to defective N-glycosylation. J Inherit Metab Dis 34:849–852
Miller BS, Freeze HH, Hoffmann GF, Sarafoglou K (2011) Pubertal development in ALG6 deficiency (congenital disorder of glycosylation type Ic). Mol Genet Metab 103:101–103
Scott K, Gadomski T, Kozicz T, Morava E (2014) Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis 37:609–617
Vuillaumier-Barrot S, Le Bizec C, Durand G, Seta N (2001) The T911C (F304S) substitution in the human ALG6 gene is a common polymorphism and not a causal mutation of CDG-Ic. J Hum Genet 46:547–548
Westphal V, Murch S, Kim S, Srikrishna G, Winchester B, Day R, Freeze HH (2000) Reduced heparan sulfate accumulation in enterocytes contributes to protein-losing enteropathy in a congenital disorder of glycosylation. Am J Pathol 157:1917–1925
Westphal V, Xiao M, Kwok PY, Freeze HH (2003) Identification of a frequent variant in ALG6, the cause of Congenital Disorder of Glycosylation-Ic. Hum Mut 22:420–421
Acknowledgments
The authors are thankful for the Euroglycanet and EURO-CDG networks
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All authors were compliant and followed the ethical guidelines, according to the requirements of JIMD
Conflict of interest
None.
Additional information
Communicated by: Marc Patterson
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Mutations described in alpha-1,3-glucosyltransferase 6 (ALG6) according to the Exome Aggregation Consortium (ExAC) database. Missense, splice site, and frameshift mutations (178 in total) were ranked by allele frequency. (The 3 or 5′ untranslated region and intronic and silent variants were filtered out.) The three common variants in our patients are highlighted. They rank 9th, 22nd, and 107th for the c.257 + 5G > A, p.A333V, and p.I299Del respectively. (XLSX 40 kb)
Rights and permissions
About this article

Cite this article
Morava, E., Tiemes, V., Thiel, C. et al. ALG6-CDG: a recognizable phenotype with epilepsy, proximal muscle weakness, ataxia and behavioral and limb anomalies. J Inherit Metab Dis 39, 713–723 (2016). https://doi.org/10.1007/s10545-016-9945-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-016-9945-x