
Abstract
Leukodystrophies are a heterogeneous group of severe genetic neurodegenerative disorders. A multiple mitochondrial dysfunctions syndrome was found in an infant presenting with a progressive leukoencephalopathy. Homozygosity mapping, whole exome sequencing, and functional studies were used to define the underlying molecular defect. Respiratory chain studies in skeletal muscle isolated from the proband revealed a combined deficiency of complexes I and II. In addition, western blotting indicated lack of protein lipoylation. The combination of these findings was suggestive for a defect in the iron-sulfur (Fe/S) protein assembly pathway. SNP array identified loss of heterozygosity in large chromosomal regions, covering the NFU1 and BOLA3, and the IBA57 and ABCB10 candidate genes, in 2p15-p11.2 and 1q31.1-q42.13, respectively. A homozygous c.436C > T (p.Arg146Trp) variant was detected in IBA57 using whole exome sequencing. Complementation studies in a HeLa cell line depleted for IBA57 showed that the mutant protein with the semi-conservative amino acid exchange was unable to restore the biochemical phenotype indicating a loss-of-function mutation of IBA57. In conclusion, defects in the Fe/S protein assembly gene IBA57 can cause autosomal recessive neurodegeneration associated with progressive leukodystrophy and fatal outcome at young age. In the affected patient, the biochemical phenotype was characterized by a defect in the respiratory chain complexes I and II and a decrease in mitochondrial protein lipoylation, both resulting from impaired assembly of Fe/S clusters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondrial encephalopathies represent a heterogeneous group of neurodegenerative disorders associated with a defect in energy metabolism. In pediatrics, most mitochondrial regressive encephalopathies are associated with lesions in basal ganglia and/or brain stem, clinically characterized by Leigh or Leigh-like syndromes (Debray et al 2007) or in the cerebral cortex in the patients presenting with Alpers syndrome (Naviaux and Nguyen 2004). Less common are mitochondrial encephalopathies associated with progressive white matter disease. The latter can result from respiratory chain deficiencies of nuclear origin as well as from defects in the mitochondrial DNA (Finsterer and Zarrouk Mahjoub 2012). Here, we report on a new form of mitochondrial leukodystrophy associated with multiple mitochondrial enzyme dysfunctions caused by a defect in the iron-sulfur cluster assembly factor IBA57 (Gelling et al 2008; Sheftel et al 2012).
The biogenesis of mitochondrial iron-sulfur (Fe/S) proteins in human cells is mediated by the highly conserved ISC (Fe/S cluster assembly) machinery (Lill 2009; Lill et al 2012). The assembly process starts with the de novo synthesis of a [2Fe-2S] cluster on the scaffold protein ISCU followed by incorporation of the cofactor into apoproteins. Both steps are assisted by numerous ISC proteins. The ISC protein IBA57 and several other maturation factors are specifically needed for maturation of mitochondrial [4Fe-4S] proteins, yet are dispensable for generation of [2Fe-2S] proteins (Sheftel et al 2012; Mühlenhoff et al 2011). [4Fe-4S] clusters are essential protein cofactors of numerous key mitochondrial enzymes, including complexes I and II of the respiratory chain, mitochondrial aconitase and lipoic acid synthase (LIAS). The latter protein generates lipoic acid, a cofactor required for the function of several enzymes like pyruvate dehydrogenase (PDH), alpha-ketoglutarate dehydrogenase (α-KGDH), and glycine cleavage system protein H. The importance of mitochondrial Fe/S proteins is reflected by the occurrence of several human diseases linked to impaired Fe/S protein biogenesis (Rouault 2012; Stehling et al 2014).
Since Fe/S proteins participate in various metabolic pathways the phenotypic outcome of the associated diseases is heterogeneous. In case of a general Fe/S protein biogenesis defect in the cell, variable phenotypes can be seen such as mitochondrial myopathy (Swedish myopathy, MIM 255125), Friedreich’s ataxia (MIM 229300), or isolated sideroblastic anemia (MIM 205950). In contrast, in case of a specific mitochondrial [4Fe-4S] protein maturation defect, severe multi-systemic diseases characterized by encephalopathy, lactic acidosis, and multiple mitochondrial dysfunctions have been reported. By now, several gene defects have been identified to be causative for such phenotypes including defects in NFU1 (MIM 608100), BOLA3 (MIM 613183), and IBA57 (MIM 615316) (Cameron et al 2011; Navarro-Sastre et al 2011; Ajit Bolar et al 2013). Interestingly, mutation of IBA57 can lead to drastically different phenotypes varying from a severe clinical phenotype with fatal outcome in the neonatal period (Ajit Bolar et al 2013), to relatively mild neurological symptoms starting in childhood (Lossos et al 2015). Here, we report yet another phenotype associated with a new mutation in IBA57 mainly characterized by fatal infantile leukodystrophy. Extensive white matter lesions with progressive neurodegeneration have already been reported in patients with other defects in the Fe/S protein maturation pathway, including in patients with NFU1 and NUBPL mutations (Nizon et al 2014; Invernizzi et al 2014; Kevelam et al 2013).
Methods
Case report
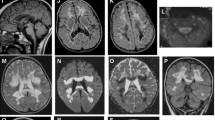
A young male patient born to consanguineous Moroccan parents presented at the age of six months with signs of motor regression due to progressive hypotonia and muscle weakness. Over a few weeks, he lost previously acquired skills, including sitting, babbling, smiling, and tracking objects. Around that age, feeding difficulties, recurrent vomiting, and feeding refusal were noticed as well as progressive irritability and crises of opisthotonus. Metabolic investigations showed a mildly increased blood lactate concentration (2.5 mmol/L, normal <2.2). Brain MRI revealed widespread white matter abnormalities in the cerebral central and periventricular areas as well as in the cerebellar white matter (Fig. 1). Lesions were also identified in the splenium of the corpus callosum, the posterior arm of the internal capsule, in the mesencephalon and the upper spinal cord. MR-spectroscopy showed an increased lactate concentration in the white matter (Fig. 1f). In CSF, the lactate concentration was increased (4.4 mmol/L; normal <2.2), and the protein concentration was normal (0.25 g/L; normal <0.45). Glycine was mildly increased in blood (460 μmol/L, normal <350) and in CSF (14 μmol/L, normal <10). Lysosomal leukodystrophies were ruled out by enzyme analysis. Signed informed consent was obtained from the parents for diagnostic procedures and molecular studies. In the following months, the patient further deteriorated. He developed a spastic tetraparesis and required nasogastric tube feeding. Ultimately, he passed away at the age of 17 months due to respiratory distress and loss of respiratory drive. Post-mortem autopsy was not performed.

Brain 1.5 Tesla MRI of the patient at age nine months. (a, b, c) Axial T2-weighted images show hyperintensities in the central and periventricular white matter, sparing subcortical U fibers. (d, e) Axial FLAIR sequences show bilaterally hyperintensities in the posterior arm of the internal capsule (d, arrows) and deep cerebellar white matter (e). (f) MR spectroscopy (TE = 135 msec) in parieto-occipital white matter revealing a lactate doublet at 1.33 ppm
Biochemical studies
Respiratory chain complex enzyme activities were measured by spectrophotometric analysis as previously described (Zecic et al 2009). In addition, respiratory chain complexes of mitochondria isolated from skeletal muscle were separated by blue native polyacrylamide gel electrophoresis, followed by staining of the enzymatic activity in the gel, as reported previously (Van Coster et al 2001). Proteins from mitochondrial fractions were also solubilized and separated by tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Western blotting was performed using a mixture of antibodies directed against one subunit in each of the respiratory chain complexes and ATP synthase, as described earlier (Ajit Bolar et al 2013). Lipoic acid-containing proteins were detected by western blotting using an antibody against protein-bound lipoic acid (ab58724, Abcam, Cambridge, UK).
Homozygosity mapping
Single-nucleotide polymorphism (SNP) genotyping was performed using the Aligent CGH+SNP 4×180 K array (G4890A-029830, Agilent Technologies, Santa Clara, CA, USA). Data were processed with Feature Extraction for CytoGenomics software and analyzed with Agilent CytoGenomics version v2.0. Homozygous regions were detected using the loss of heterozygosity (LOH) algorithm at the default threshold of 6.0. Filtering of AOH regions was carried out using the BENCHlab CNV software (Cartagenia, Leuven, Belgium).
Exome sequencing and analysis
In brief, exon capture was achieved using the SureSelect Human V4 (51 Mb) Kit. Subsequently, exon-enriched DNA fragments were loaded on the HiSeq2000 System (Illumina, Santiago, CA) for paired-end sequencing. The mean exome coverage was set as 50-fold. Raw sequence data were aligned to the human reference genome (build hg19) by Burrows-Wheeler Aligner, and sequence variants called using GATK (http://www.broadinstitute.org/gatk/).Variants analysis was perform with a BENCHlab NGS (Cartagenia, Inc.) based pipeline, including candidate gene list variant filtering, variant reviewing for frequency in dbSNP (http://www.ncbi.nlm.nih.gov/snp/) and 1000 genomes (http://www.1000genomes.org/), and variant filtering by conservation score (PhyloP). The predicted effect on protein function was analyzed using SIFT (http://sift.jcvi.org) and Mutationtaster (http://www.mutationtaster.org/).
Site-directed in vitro mutagenesis
Wild type IBA57 cDNA was subcloned in plasmid pEGFP-N1 (Clonetech) as previously reported (Ajit Bolar et al 2013). To introduce the mutation, a synthetic oligonucleotide containing IBA57 c.436C > T modification was cloned between restriction sites BglII-XmaI (GeneScript). For cell biological studies IBA57 and IBA57_R146W were subcloned into pEGFP-N1_ Δ1-520 to exchange the enhancer/promoter cassette of pEGFP-N1 for a promoter that produces roughly wild-type levels of IBA57 protein (Nalaskowski et al 2012).
Functional studies
To examine the pathogenicity of the IBA57 variant HeLa cells were depleted for IBA57 protein using an RNAi approach, as reported previously (Ajit Bolar et al 2013). Briefly, HeLa cells were transfected thrice with specific siRNAs against IBA57 at a 3 day interval. A fraction of cells were co-transfected with equal amounts of expression plasmid encoding either wild-type IBA57 or the mutant version (IBA57_R146W). Nine days after initial transfection cells were harvested and analyzed for enzyme activities by spectrophotometric analysis and protein abundance by western blot, as reported earlier (Ajit Bolar et al 2013). Experiments were performed in triplicate. Immunoblots were quantified by densitometry and presented relative to the mitochondrial F1α/β ATP synthase expression.
Results
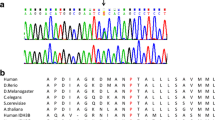
Respiratory chain enzyme activity studies in skeletal muscle revealed a combined deficiency of complexes I and II (Table 1) suggesting a possible Fe/S cluster biosynthesis defect. BN-PAGE and western blotting using a mixture of antibodies directed against one subunit in each of the oxidative phosphorylation complexes showed a strong reduction of complexes I and II, and a moderate decline of complex IV (Fig. 2a, b). Analysis of the lipoylation of the E2 subunits of pyruvate dehydrogenase (PDH) and alpha-ketoglutarate dehydrogenase (αKGDH) revealed a severe decrease of this cofactor for both enzymes (Fig. 2c), providing a further hint for disturbance of the Fe/S cluster assembly pathway. SNP array identified loss of heterozygosity in large chromosomal regions, covering the NFU1 and BOLA3, and the IBA57 and ABCB10 candidate genes, in 2p15-p11.2 and 1q31.1-q42.13, respectively. Using whole exome sequencing a homozygous mutation c.436C > T (p.Arg146Trp) was detected in IBA57 which was confirmed by Sanger sequencing. Both parents were heterozygous for the mutation. Arginine 146 is conserved in most species, even though the IBA57 sequence encompasses only a 40 amino acid residue long segment of higher conservation (Ajit Bolar et al 2013). No mutation was found in the NFU1, BOLA3, and ABCB10 genes, both in exome data and by Sanger sequencing.

Diminution of the protein levels and activities of respiratory chain complexes I and II and of lipoic acid-dependent enzymes in IBA57 patient cells. (a) Western blot showing expression levels for one subunit of each respiratory chain complex and ATP synthase (NDUFB8 for complex I, Ip for complex II, core2 for complex III, COXII for complex IV and V alpha for ATP synthase) in skeletal muscle of patient (P) and controls (C1, C2). To illustrate the degree of decrease in protein expression a control was loaded at 25 % (C2 25 %). (b) The in-gel activities of different respiratory chain complexes were measured in extracts from skeletal muscle mitochondria of patient (P) and control (C) cells using BN-PAGE. Patient cells show severely decreased activities for complexes I and II, a slight reduction of complex IV, whereas complexes III and V were normal. (c) Western blot illustrating the faulty lipoylation on PDH-E2 and αKGDH-E2 in patient (P) compared to control cells (C1, C2). To illustrate the degree of the decrease a control was loaded at 25 % (C2 25 %). Porin was used as loading control. (d) Western blot illustrating the close to normal IBA57 expression in patient’s (P) skeletal muscle and in cultured skin fibroblasts compared to controls (C1, C2, C3). V alpha was used as loading control
The mutated IBA57 protein was present at normal levels in cultured skin fibroblasts from the proband, and slightly decreased in skeletal muscle (Fig. 2d). We thus suspected that the amino acid exchange caused a functional disturbance rather than proteolytic sensitivity of IBA57. To test this assumption, HeLa cells were depleted for IBA57 by RNAi technology (Sheftel et al 2012; Ajit Bolar et al 2013). We analyzed the ability of plasmid-encoded wild-type IBA57 and mutant IBA57 (IBA57_R146W) to complement the deficiency of the native protein. To avoid overproduction of plasmid-encoded proteins, a series of pEGFP-N1 plasmids were used containing truncated versions of the enhancer/promoter cassette. The version that allowed production of nearly wild-type levels of IBA57 or IBA57_R146W proteins was used for further studies (Supplementary Fig. 1). The equal amounts of plasmid-produced wild-type and mutant IBA57 proteins demonstrated that the mutation did not significantly affect the stability of IBA57, unlike a previously described amino acid exchange (Ajit Bolar et al 2013). HeLa cells depleted for IBA57 showed a characteristic strong depletion of the [4Fe-4S] cluster-containing respiratory chain complexes I and II (SDH), and of the lipoic acid levels of PDH and αKGDH, two enzymes dependent on the [4Fe-4S] protein LIAS (Sheftel et al 2012)(Fig. 3). In addition, the non-Fe/S complex IV (COX) was strongly decreased which is known to be an indirect effect of IBA57 defects (Gelling et al 2008; Sheftel et al 2012; Ajit Bolar et al 2013). These enzyme (Fig. 3a) or protein level (Fig. 3b and c) deficiencies could be (partially) restored by expression of wild-type IBA57. In contrast, mutant IBA57_R146W was not able to significantly improve the depletion phenotype. As a control, mitochondrial complex V (F1Fo ATP synthase), the [2Fe-2S] cluster-containing mitochondrial ferrochelatase or the cytosolic [4Fe-4S] protein GPAT were not significantly affected (Fig. 3b and c). In conclusion, the amino acid exchange R146W in IBA57 impairs its function in mitochondrial [4Fe-4S] protein biogenesis without affecting its stability.

The major targets of IBA57 deficiency can be functionally restored by wild-type but not mutant IBA57. HeLa cells were transfected with IBA57-specific siRNA thrice at a three day interval. Cells were additionally co-transfected with plasmids containing no gene, wild-type or mutant IBA57 (IBA57_R146W). Cells were analyzed nine days after initial transfection. (a) Enzyme activities of mitochondrial respiratory complexes II (SDH) or IV (COX) were measured spectrophotometrically. Values were normalized to the activity of mitochondrial citrate synthase, and are presented relative to the activities of mock-transfected control cells. In contrast to wild-type IBA57, IBA57_R146W was unable to revert SDH and COX activities. (b) Western blot analysis of cell extracts from part A. Antibodies were used against mitochondrial IBA57, NDUFA9 (complex I), SDHB (complex II), COX2 (complex IV), F1α/β (complex V), lipoic acid (LA) of PDH-E2 or αKGDH-E2, ferrochelatase (FeCh) and cytosolic glutamate phosphoribosylpyrophosphate amidotransferase (GPAT). Restoration of relevant protein levels is observed only upon synthesis of wild-type but not mutant IBA57. Representative immunoblots are shown. (c) Densitometric quantitation of the data shown in part B. Data are presented as mean ± SD (n = 3)
Discussion
IBA57 deficiency was first reported in two siblings who were already severely ill at birth and who were found to have generalized muscle weakness, dysmorphic features, arthrogryposis, lactic acidosis and hyperglycinemia, leading to early death. Brain MRI predominantly showed frontoparietal polymicrogyria, hypoplasia of the corpus callosum and the medulla oblongata (Ajit Bolar et al 2013). In contrast, a phenotypically rather mild IBA57 hypomorphic mutation was recently reported in association with a slowly progressive childhood-onset neurological appearance characterized by symptoms of a rare form of spastic paraparesis, optic atrophy and peripheral neuropathy (SPOAN) (Lossos et al 2015). The IBA57 patient presented here showed an intermediate phenotype. He was asymptomatic at birth, and only at the age of six months presented signs of regressive encephalopathy, eventually leading to death one year later. Unlike the patients with the neonatal form of IBA57 deficiency, brain MRI of the proband did not display cerebral cortical malformation but showed obvious signs of leukodystrophy. The clinical course in the proband was reminiscent of that reported in the patients harboring pathogenic mutations in the NFU1 gene, another late-acting targeting factor of the Fe/S cluster assembly pathway (Cameron et al 2011; Navarro-Sastre et al 2011), in whom white matter anomalies have also been observed, including true leukodystrophy (Nizon et al 2014; Invernizzi et al 2014). Most of these patients were asymptomatic at birth and presented with neurological regression starting at age 1–9 months, frequently associated with failure to thrive and pulmonary hypertension, usually leading to death before the age of 2 years. Progressive leukoencephalopathy with neurological regression and lactate increase has also been reported in patients with mutations in NUBPL, another Fe-S assembly factor specifically implicated in respiratory complex I biogenesis. However in these cases, patients harbored isolated complex I deficiency and normal activity of lipoylated mitochondrial enzymes. In addition, a specific pattern of brain MRI anomalies has been recognized in NUBPL related diseases, including predominant involvement of the cerebellar cortex, deep cerebral white matter and corpus callosum (Kevelam et al 2013).
Glycine concentration is typically increased in CSF from the patients with NFU1-related disease and also was significantly increased in the patients with the neonatal form of IBA57 deficiency (Ajit Bolar et al 2013). In contrast, the patients presenting with the SPOAN phenotype had normal glycine concentration in CSF (Lossos et al 2015). In the patient described here, despite clinical and radiological signs of severe leukodystrophy, glycine was only mildly increased in blood and CSF, indicating that multiple mitochondrial dysfunction syndrome (MMDS) should be considered even in the absence of a severe glycine increase. Altogether, the phenotypic spectrum of IBA57-related pathologies suggests a continuum of severity in clinical signs, biological markers and biochemical defects.
The mutation identified here in IBA57 was associated with normal amounts of IBA57 protein in patient’s tissues. However, the mutant protein appeared to be hardly effective in Fe/S protein biogenesis, as shown by the biochemical studies in patient tissues revealing that both the levels and activities of several [4Fe-4S] cluster-containing enzymes were impacted by this mutation. Most affected were the respiratory chain complexes I and II, as well as the lipoic acid levels of the E2 subunits of PDH and α-KGDH indicative of a defect in the Fe/S protein LIAS. Moreover, the Fe/S cluster-independent complex IV (COX) was affected, similarly as in yeast or cell culture models of IBA57 depletion (Gelling et al 2008; Sheftel et al 2012). The mechanistic biochemical explanation of this latter observation remains to be resolved, but notably mutations in NFU1 or BOLA3 do not affect COX activities (Cameron et al 2011; Navarro-Sastre et al 2011). In contrast, the level of a subunit of the [2Fe-2S] cluster-dependent complex III was unaffected supporting the idea that IBA57 is specific for maturation of [4Fe-4S] proteins. All these biochemical findings in patient’s tissues are consistent with those detected in the siblings with severe IBA57 deficiency (Ajit Bolar et al 2013). The observations made on patient material were verified in human cell culture where IBA57 was depleted by RNAi technology. Here, expression of wild-type but not mutant IBA57 was able to (partially) complement the defects in complex II, COX, PDH, and α-KGDH levels and/or enzyme activities. In other cases, the signal to noise ratio was too small to be significant. The slight complementation observed for lipoic acid levels attached to E2-PDH may suggest some weak residual function of mutant IBA57. This also may explain the late onset of the disease, and possibly the absence of a severe increase of glycine concentrations in serum and CSF.
The IBA57 mutation reported here leads to an exchange of residue Arg146 to Trp. IBA57 is a poorly conserved protein which shows only a stretch of 40 residues with strong conservation (Ajit Bolar et al 2013). The region around residue Arg146 does not belong to this conserved area. Nevertheless, Arg146 itself is conserved in the majority of species including some bacterial proteins. From a crystal structure of the homologous YgfZ protein from E. coli the mutation is predicted to be surface-exposed and located at the end of an α-helix in front of a turn leading into a β-sheet structure (Teplyakov et al 2004). However, the E. coli protein contains a phenylalanine residue instead of arginine in this position thus precluding mechanistic predictions of how the R146W mutation may impair protein functionality. Nevertheless, it is possible to estimate the degree of functional impairment of IBA57_R146W from a comparison of the current case with two previously described mutations which do not impair protein function but rather severely decrease the IBA57 protein levels (Ajit Bolar et al 2013; Lossos et al 2015). In these cases, hardly any or approximately 10 % residual IBA57 is correlated with the fatal neonatal or relatively mild neurological phenotypes, respectively, as well as with the residual biochemical activities of Fe/S cluster-dependent enzymes. The intermediate severity of the current case hence suggests that the function of IBA57 was at least 20-fold decreased by the R146W mutation.
In conclusion, a pathogenic mutation in IBA57 can cause severe autosomal recessive neurodegeneration clinically characterized by a severe and rapidly progressive leukodystrophy. Biochemically, it is characterized by a complex biochemical phenotype, including a specific pattern of combined respiratory chain complex deficiency and a defect in mitochondrial protein lipoylation, both caused by the primary impairment of the Fe/S protein biogenesis pathway.
References
Ajit Bolar N, Vanlander AV, Wilbrecht C et al (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet 22:2590–2602
Cameron JM, Janer A, Levandovskiy V et al (2011) Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet 89:486–495
Debray FG, Lambert M, Chevalier I et al (2007) Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 119:722–733
Finsterer J, Zarrouk Mahjoub S (2012) Leukoencephalopathies in mitochondrial disorders: clinical and MRI findings. J Neuroimaging 22:e1–e11
Gelling C, Dawes IW, Richhardt N, Lill R, Mühlenhoff U (2008) Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes. Mol Cell Biol 28:1851–1861
Invernizzi F, Ardissone A, Lamantea E et al (2014) Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutation. Front Genet 5:412. doi:10.3389/fgene.2014.00412
Kevelam SH, Rodenburg RJ, Wolf NI et al (2013) NUBPL mutations in patients with complex I deficiency and a distinct MRI pattern. Neurology 80:1577–1583
Lill R (2009) Function and biogenesis of iron-sulfur proteins. Nature 460:831–838
Lill R, Hoffmann B, Molik S et al (2012) The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochem Biophys Acta 1823:1491–1508
Lossos A, Stümpfig C, Stevanin G et al (2015) Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology 84:659–667
Mühlenhoff U, Richter N, Pines O, Pierik AJ, Lill R (2011) Specialized function of yeast Isa1 and Isa2 proteins in the maturation of mitochondrial [4Fe-4S] proteins. J Biol Chem 286:41205–41216
Nalaskowski MM, Ehm P, Giehler S, Mayr GW (2012) A toolkit for graded expression of green fluorescent protein fusion proteins in mammalian cells. Anal Biochem 428:24–27
Navarro-Sastre A, Tort F, Stehling O et al (2011) A fatal mitocondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet 89:656–667
Naviaux RK, Nguyen KV (2004) POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol 55:706–712
Nizon M, Boutron A, Boddaert N et al (2014) Leukoencephalopathy with cysts and hyperglycinemia may result from NFU1 deficiency. Mitochondrion 15:59–64
Rouault TA (2012) Biogenesis of iron-sulfur clusters in mammalain cells: new insights and relevance to human disease. Dis Model Mech 5:155–164
Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsasser HP, Mühlenhoff U, Lill R (2012) The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol Biol Cell 23:1157–1166
Stehling O, Wilbrecht C, Lill R (2014) Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie 100:61–77
Teplyakov A, Obmolova G, Sarikaya E et al (2004) Crystal structure of the YgfZ protein from Escherichia coli suggests a folate-dependent regulatory role in one-carbon metabolism. J Bacteriol 186:7134–7140
Van Coster R, Smet J, George E et al (2001) Blue native polyacrylamide gel electrophoresis: a powerful tool in diagnosis of oxidative phosphorylation defects. Pediatr Res 50:658–665
Zecic A, Smet J, De Praeter CM et al (2009) Lactic acidosis in a newborn with adrenal calcifications. Pediatr Res 66:317–322
Acknowledgments
This work was supported by a Fund Invest for Scientific Research (FIRS) (grant number 4709) from the Centre Hospitalier Universitaire de Liège, Belgium. We gratefully acknowledge the donation of the promotor-truncated versions of plasmid pEGFP-N1 from Dr. G.W. Mayr, Hamburg. RL acknowledges generous financial support from Deutsche Forschungsgemeinschaft (SFB 593, GRK 1216, and SPP 1710), and the LOEWE program of state Hessen. This work was also supported by the Special Research Fund (BOF) from the Ghent University (grant number B/00559/0).
Compliance with ethics guidelines
ᅟ
Conflict of interest
None.
Informed consent
All procedures followed were in accordance with the ethical standards of the responsiblecommittee on human experimentation and with the Helsinki Declaration of 1975, as revised in2000. Written informed consent was obtained for patients being included in this study.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by: Jerry Vockley
François-Guillaume Debray, Claudia Stümpfig and Arnaud V. Vanlander contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(JPEG 123 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Debray, FG., Stümpfig, C., Vanlander, A.V. et al. Mutation of the iron-sulfur cluster assembly gene IBA57 causes fatal infantile leukodystrophy. J Inherit Metab Dis 38, 1147–1153 (2015). https://doi.org/10.1007/s10545-015-9857-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9857-1