
Abstract
Targeting lysosomal enzymes to receptors involved in transport into and across cells holds promise to enhance peripheral and brain delivery of enzyme replacement therapies (ERTs) for lysosomal storage disorders. Receptors being explored include those associated with clathrin-mediated pathways, yet other pathways seem also viable. Well characterized examples are that of transferrin receptor (TfR) and intercellular adhesion molecule 1 (ICAM-1), involved in iron transport and leukocyte extravasation, respectively. TfR and ICAM-1 support ERT delivery via clathrin- vs. cell adhesion molecule-mediated mechanisms, displaying different valency and size restrictions. To comparatively assess this, we used antibodies vs. larger multivalent antibody-coated carriers and evaluated TfR vs. ICAM-1 binding and endocytosis in endothelial cells, as well as in vivo biodistribution and delivery of a model lysosomal enzyme required in peripheral organs and brain: acid sphingomyelinase (ASM), deficient in types A-B Niemann Pick disease. We found similar binding of antibodies to both receptors under control conditions, with enhanced binding to activated endothelium for ICAM-1, yet only anti-TfR induced endocytosis efficiently. Contrarily, antibody-coated carriers showed enhanced binding, engulfment, and endocytosis for ICAM-1. In mice, anti-TfR enhanced brain targeting over anti-ICAM, with an opposite outcome in the lungs, while carriers enhanced ICAM-1 targeting over TfR in both organs. Both targeted carriers enhanced ASM delivery to the brain and lungs vs. free ASM, with greater enhancement for anti-ICAM carriers. Therefore, targeting TfR or ICAM-1 improves lysosomal enzyme delivery. Yet, TfR targeting may be more efficient for smaller conjugates or fusion proteins, while ICAM-1 targeting seems superior for multivalent carrier formulations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
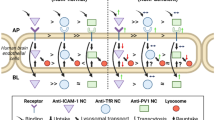
The lysosomal storage disorders (LSDs) are rare diseases mainly arising from genetic defects affecting lysosomal enzymes, and typically cause dysfunction in peripheral organs and the central nervous system (CNS) (Futerman and van Meer 2004). Enzyme replacement therapy (ERT) is a viable treatment for LSDs, yet suboptimal delivery limits this approach (Brady 2003; Desnick and Schuchman 2002). For example, in peripheral tissues excluding the reticuloendothelial system (RES) in liver and spleen, continuous endothelial cells (ECs) lining the microcirculation limit enzyme transport into the tissue parenchyma (Pardridge and Boado 2012; Schnitzer 2001). CNS penetration is particularly difficult as the blood–brain barrier (BBB) greatly restricts paracellular transport (i.e., between adjacent ECs), and the transcellular route is mainly limited to clathrin-mediated endocytosis (Begley et al 2008; Banks 2009; Pardridge and Boado 2012). Inadequate glycosylation of recombinant lysosomal enzymes, along with impaired expression and/or clathrin-mediated endocytosis via mannose-6-phosphate (M6P) receptor in some LSDs, pose additional obstacles for ERT (Cardone et al 2008; Dhami and Schuchman 2004; Mistry et al 1996). Additionally, BBB transport is impaired by downregulation of M6P receptor after birth (Urayama et al 2004).
A promising strategy to enhance ERT is glycosylation-independent targeting for transport across endothelium and into lysosomes within tissue cells. Several strategies have been explored, including targeting with HIV Tat peptides (Vaags et al 2005; Xia et al 2001; Zhang et al 2008), insulin growth factor II (LeBowitz et al 2004), receptor associated protein RAP (Prince et al 2004), or by targeting the insulin receptor (Boado et al 2008; Lu et al 2011), transferrin receptor (TfR) (Boado et al 2009, 2011; Osborn et al 2008; Zhou et al 2012; Xia et al 2000; Chen et al 2008), or intercellular adhesion molecule 1 (ICAM-1) (Muro et al 2006a; Garnacho et al 2008a; Hsu et al 2011, 2012). While Tat peptides provide targeting via non-specific charge-mediated interaction, targeting cell surface receptors involves association with particular endocytic transport mechanisms, e.g., cell adhesion molecule- (CAM)-mediated transport for ICAM-1 or clathrin-mediated transport for all other strategies (Muro 2010). Among clathrin-mediated strategies, targeting TfR is particularly well studied.
TfR is a transmembrane glycoprotein expressed on the surface of many cells, including brain capillary endothelium (Pardridge 2010; Jefferies et al 1984). TfR enables iron transport across cellular barriers via transcytosis (e.g., in the BBB) and into cells by clathrin-mediated endocytosis (Conrad and Umbreit 2000; Dautry-Varsat 1986; Fishman et al 1987). This process involves formation of ∼100–150 nm clathrin-coated pits, where engaged receptors interact with cytosolic adaptor proteins which bind clathrin triskelia, leading to formation of a polyhedral protein lattice around the invaginating vesicle (Hirst and Robinson 1998; Steven et al 1983). Concerted action of dynamin and the actin cytoskeleton helps pinch off clathrin-coated pits into the cytosol, with subsequent microtubular-mediated transport (Jin and Snider 1993). This pathway is induced by engagement of TfR with transferrin, and other “ligands” such as antibodies, peptides and aptamers (Boado et al 2009, 2011; Osborn et al 2008; Zhou et al 2012; Xia et al 2000; Chen et al 2008), or drug delivery carriers displaying these affinity moieties (Ko et al 2009; Pang et al 2011; Shi et al 2001).
ICAM-1 is another transmembrane glycoprotein expressed on ECs (including the BBB) and most other cell types (Rothlein et al 1986; Marlin and Springer 1987). ICAM-1 is a co-receptor for β2 integrins, helping in adhesion and extravasation of leukocytes during inflammation (Rothlein et al 1986; Marlin and Springer 1987). ICAM-1 is not an endocytic receptor per se, since no soluble endocytic ligands are known for this molecule, and antibodies binding ICAM-1 are poorly internalized (Murciano et al 2003). However, cell binding of conjugates or drug carriers displaying multiple ICAM-1 targeting antibodies or peptides enables uptake within and across cells via the CAM-mediated pathway, a route different from classical clathrin or caveolar endocytosis, macropinocytosis, and phagocytosis (Muro et al 2003; Garnacho et al 2012; Ghaffarian et al 2012). CAM endocytic vesicles have no protein coat. However, signaling through the sphingomyelin/ceramide cascade and interaction of ICAM-1 with sodium/proton exchanger protein 1 (NHE1) results in local physicochemical changes at the plasma membrane and re-organization of the actin cytoskeleton, leading to effective uptake of ICAM-1-targeted carriers up to several micrometers in size (Muro et al 2006b, 2008; Serrano et al 2012).
Hence, different valency and size requirements may exist for uptake of materials via CAM- vs. clathrin-mediated endocytosis. This is relevant to development of targeting strategies for therapeutics utilizing smaller, monovalent fusion proteins or conjugates versus larger, multivalent conjugates or drug carriers. However, delivery through these routes has not been assessed comparatively. We used fluorescence microscopy and radioisotope tracing to investigate the behavior of antibodies and antibody-coated carriers targeted to ICAM-1 vs. TfR in terms of binding and endocytosis in ECs, as well as biodistribution and lysosomal enzyme delivery in mice.
Methods
Antibodies and reagents
Monoclonal antibodies against human and murine ICAM-1 were R6.5 (Marlin and Springer 1987) and YN1 (Jevnikar et al 1990), respectively. Monoclonal antibodies against human and murine TfR were T56/14 from EMD Millipore (Billerica, MA) and 8D3 from Novus Biologicals (Littleton, CO). ICAM-1-targeting γ3-derivative peptide, NNQKIVNIKEKVAQIEA (Altieri et al 1995; Garnacho et al 2012), was synthesized by United Biochemical Research (Seattle, WA). Transferrin and secondary antibodies were from Molecular Probes (Eugene, OR). Recombinant human ASM was produced in chinese hamster ovary cells and purified as described (He et al 1999). Polystyrene particles (115 nm, 1 μm or 4.5 μm) were from Polysciences (Warrington, PA). Na125I and iodobeads were respectively from Perkin Elmer (Wellesley, MA) and Thermo Scientific (Rockford, IL). Remaining reagents were from Sigma Chemicals (St. Louis, MO).
Preparation and characterization of carriers targeted to ICAM-1 or TfR
Model polymer carriers were prepared by surface adsorption as described (Calderon et al 2011; Muro et al 2008; Garnacho et al 2012). For cell culture experiments, polystyrene particles (115 nm, 1 μm, or 4.5 μm) were coated with antibodies (anti-ICAM, anti-TfR, or control IgG) or alternative ligands (ICAM-1-binding peptide (γ3-derivative) or transferrin). For experiments in mice, anti-ICAM, anti-TfR, or IgG carriers contained additionally 125I-IgG as a tracer (95:5 antibody mass ratio) or 125I-ASM as model lysosomal enzyme cargo (50:50 antibody to enzyme mass ratio). Alternatively, 125I-anti-ICAM or 125I-anti-TfR was used to determine the number of antibodies coated per carrier (valency). Uncoated materials were removed by centrifugation, and carriers were resuspended in phosphate buffered saline supplemented with 1 % bovine serum albumin and sonicated to avoid aggregation. Final carrier size, polydispersity, and zeta potential were estimated using a Malvern Zetasizer (Table 1).
Cell cultures
Pooled human umbilical vein endothelial cells (HUVECs) (Lonza Walkersville, Inc., Walkersville, MD) were seeded onto gelatin-coated coverslips, and cultured at 37 °C, 5 % CO2, and 95 % relative humidity. Cells (3–4 passages) were grown in M-199 medium supplemented with 15 % fetal bovine serum, 2 mM glutamine, 15 μg/ml endothelial cell growth supplement, 100 μg/ml heparin, 100 μg/ml penicillin, and 100 μg/ml streptomycin. When indicated, inflammatory condition was mimicked by 16–20 h TNFα pre-treatment.
Binding of antibodies and antibody-coated carriers to endothelial cells
Control or TNFα-activated HUVECs were incubated for 15 min or 1 h with free anti-ICAM or anti-TfR, or FITC-labeled carriers coated with anti-ICAM, anti-TfR, γ3, or transferrin (∼250 nm, 1 μm, or 4.5 μm final diameter). After removing unbound materials, cells were fixed with 2 % paraformaldehyde. Free antibodies were visualized by staining with FITC-labeled goat anti-mouse IgG, while antibody-coated carriers contained FITC within the polymer matrix. Fluorescence microscopy images were taken using an Olympus IX81 microscope (Olympus, Inc., Center Valley, PA), ORCA-ER camera (Hamamatsu, Bridgewater, NJ), 60× objective (Olympus Uplan F LN; Olympus) and FITC-optimized filter (3540B-OMF; Semrock, Inc., Rochester, NY). Images were acquired with SlideBook 4.2 (Intelligent Imaging Innovations, Denver, CO), and analyzed using Image-Pro 6.3 (Media Cybernetics, Inc., Bethesda, MD) to estimate antibody binding by mean fluorescence intensity, or carriers bound per cell by counting the number of fluorescent objects. Cell borders were delimited by phase-contrast.
Imaging endothelial engulfment of antibody-coated carriers
To examine initial stages of carrier engulfment, HUVECs were incubated for 15 min at 37 °C with anti-ICAM or anti-TfR carriers. Particles were 4.5 μm to allow detailed visualization of engulfment structures (Serrano et al 2012). After washing unbound carriers, cells were fixed, permeabilized with 0.2 % Triton X-100, and immunolabeled to detect enrichment around bound carriers of NHE1 or clathrin heavy chain, respectively (Serrano et al 2012).
Endocytosis of antibodies and antibody-coated carriers by endothelial cells
Control or TNFα-activated HUVECs were incubated at 37 °C for 1 h with free anti-ICAM or anti-TfR, or ∼250 nm FITC-labeled anti-ICAM or anti-TfR carriers. After washing to remove unbound materials, cells were fixed, and surface-bound materials were stained using Texas-Red-labeled goat anti-mouse IgG. For free antibodies, cells were subsequently permeabilized and incubated with FITC-labeled secondary antibody which stained both surface + internalized primary antibody. Fluorescence microscopy was used to distinguish internalized materials as FITC single-labeled antibodies or carriers, from surface-bound materials appearing yellow due to Texas-Red + FITC double-labeling. The percentage internalization was calculated from images as described (Muro et al 2003).
Biodistribution and lysosomal enzyme delivery by antibodies and antibody-coated carriers
Anesthetized C57BL/6J mice were injected intravenously with 125I-labeled anti-ICAM, anti-TfR, or control IgG as either free or ∼250 nm carrier-coated counterparts (∼1.3 mg antibody/kg body weight, ∼1.8 × 1013 particles/kg). Alternatively, mice were injected with 125I-ASM as free counterpart or coated on ∼250 nm anti-ICAM or anti-TfR carriers (∼0.7 mg ASM/kg body weight, ∼1.8 × 1013 particles/kg). Blood samples were collected from the retro-orbital plexus at 1, 15, and 30 min after injection, and organs (brain, lungs, and liver) were harvested at 30 min. 125I content and weight of samples were determined to estimate the specificity index (SI), calculated as the localization ratio (LR) of targeted formulations divided by non-targeted counterparts (IgG, IgG carriers, or free ASM). LR is the percent injected dose per gram of tissue divided by percent injected dose per gram of blood. Hence, SI reflects specific targeting to organs, normalized for organ size and blood fraction of the formulation (Hsu et al 2011). Studies followed IACUC and University of Maryland regulations.
Statistics
Data were calculated as mean ± S.E.M, where statistical significance was determined as p ≤ 0.05 by Student’s t-test.
Results
Binding of antibodies and antibody-coated carriers targeted to ICAM-1 vs. TfR on endothelial cells
Targeting therapeutics or drug carriers to ICAM-1 or TfR enhances accumulation in the body, such as in ECs controlling transport from the circulation to tissues (e.g., brain). Yet, the comparative efficiency of these targeting strategies has not been addressed.
We first analyzed binding of free targeting moieties (monoclonal antibodies) to ICAM-1 vs. TfR on ECs in control or inflammatory-like conditions (relevant in numerous disease states, including LSDs). Fluorescence microscopy showed similar binding of anti-TfR or anti-ICAM under control conditions (Fig. 1a and Suppl. Fig. 1). However, in agreement with ICAM-1 overexpression during inflammation (Rothlein et al 1986; Marlin and Springer 1987), anti-ICAM binding increased markedly in ECs pre-treated with TNFα (15-fold), whereas anti-TfR increased modestly (1.6-fold). Consequently, bound anti-ICAM greatly exceeded (by 9.6-fold) anti-TfR under inflammatory stimulation.

Binding of antibody and antibody-coated carriers targeted to ICAM-1 vs. TfR on endothelial cells. a Binding of antibodies to ICAM-1 vs. TfR was tested by immunofluorescence after incubation for 15 min at 37 °C with control or TNFα-activated HUVECs. b Binding of ∼250 nm FITC-labeled anti-ICAM vs. anti-TfR carriers was assessed by fluorescence microscopy after incubation for 1 h with fixed (control or TNFα-activated) HUVECs. Data are mean ± SEM, with n ≥ 25 cells. * compares control vs. TNFα for each target, and # compares ICAM-1 vs. TfR for each condition. * or # represent p ≤ 0.05, ** or ## represent p ≤ 0.01, and *** or ### represent p ≤ 0.001, by Student's t-test
We next compared binding of antibody-coated carriers. Anti-ICAM and anti-TfR carriers displayed similar size (∼250 nm), polydispersity (∼0.180), zeta potential (∼−9 mV), and valency (∼275–300 antibodies/carrier particle).
Despite similar antibody binding under control conditions, anti-ICAM carriers displayed two-fold enhanced binding to ECs (Fig. 1b and Suppl. Fig. 2). This also occurred for carriers coated with smaller affinity moieties, namely γ3-derivative peptide vs. transferrin (8.6-fold enhancement; data not shown). For activated ECs, binding of anti-ICAM carriers was enhanced further compared to anti-TfR carriers (17.4-fold difference). Hence, ICAM-1-targeted carriers enhanced binding by eight-fold in inflammatory-like condition, whereas binding of TfR-targeted carriers was unchanged compared to control conditions. Similar behavior was observed with larger carriers (Fig. 2). For instance, 1 μm anti-ICAM carriers displayed ∼2.1-fold increased binding over anti-TfR carriers in control ECs (Fig. 2a), and ∼8.4-fold increased binding in TNFα-activated cells. Binding of anti-ICAM carriers in inflammatory conditions remained enhanced (∼2.3 fold) for 4.5 μm carriers (Fig. 2b). Therefore, targeting ICAM-1 seems favored for EC binding, particularly when utilizing carriers.

Endothelial binding of anti-ICAM-1 vs. anti-TfR carriers of different size. a Binding of ∼1 μm anti-ICAM vs. anti-TfR carriers to control or TNFα-activated HUVECs was assessed by fluorescence microscopy after 15 min incubation at 37 °C as in Fig. 1. b Binding after 15 min incubation at 37 °C with TNFα-activated HUVECs was comparatively assessed in the case of ∼250 nm, ∼1 μm, or ∼4.5 μm anti-ICAM vs. anti-TfR carriers. Data are mean ± SEM, with n ≥ 75 cells. * compares control vs. TNFα for each target, and # compares anti-ICAM vs. anti-TfR for each carrier size. *** or ### represent p ≤ 0.001, by Students t-test
Endothelial endocytosis of antibodies and antibody-coated carriers targeted to ICAM-1 vs. TfR
Endocytic internalization is critical for successful transport of therapeutics into and across ECs. Hence, we next compared endocytosis of antibodies or antibody-coated carriers.
Confirming previous (yet not comparative) observations, we found anti-ICAM was poorly internalized by ECs compared with anti-TfR (Fig. 3a and Suppl. Fig. 3), even with TNFα activation where anti-ICAM binding greatly exceeded that of anti-TfR. Contrarily, internalization of ICAM-1-targeted carriers exceeded TfR-targeted carriers (Fig. 3b, c and Suppl. Fig. 4) in control and with TNFα activation, suggesting more efficient carrier uptake by CAM-mediated vs. clathrin-mediated endocytosis. Due to enhanced binding and uptake, the absolute amount of internalized carriers was ∼2.6-fold greater when targeting ICAM-1 vs. TfR.

Endothelial endocytosis of antibody and antibody-coated carriers targeted to ICAM-1 vs. TfR. a Uptake of anti-ICAM vs. anti-TfR was assessed after 1 h incubation at 37 °C with TNFα-activated HUVECs. This was done by fluorescence analysis after removing unbound antibodies, followed by staining surface-bound antibodies with a Texas-Red secondary IgG, cell permeabilization, and finally staining both surface + internalized antibodies with a FITC-labeled secondary IgG. Percentage of internalization was calculated as the fraction of FITC single-labeled antibody compared to total cell-associated antibody (single-labeled in FITC + double-labeled in FITC and Texas-Red). b–c Uptake of ∼250 nm FITC-labeled anti-ICAM vs. anti-TfR carriers was assessed after 1 h incubation at 37 °C with control or TNFα-activated HUVECs. Unbound carriers were removed and surface-bound carriers were stained with a Texas-Red secondary IgG. Internalization was quantified as described for antibodies, and expressed as percentage of uptake compared to total cell-associated carriers (b) or absolute number of carriers internalized per cell (c). Data are mean ± SEM, with n ≥ 15 cells. * compares control vs. TNFα for each target, and # compares anti-ICAM vs. anti-TfR carriers. *** or ### represents p ≤ 0.001, by Students t-test
We then visualized formation of membrane engulfment structures around carriers bound to ECs and recruitment of molecular partners associated to CAM vs. clathrin pathways. Large 4.5 μm carriers were used to facilitate immunofluorescent imaging of NHE1 or clathrin heavy chain enrichment at binding sites of anti-ICAM or anti-TfR carriers. As shown in Fig. 4, ICAM-1 binding lead to rapid (within 15 min) formation of NHE1-enriched engulfment structures at the plasmalemma, while engulfment structures enriched in clathrin heavy chain were much less apparent for anti-TfR carriers. This result pairs well with greater vesicular endocytosis observed for anti-ICAM carriers.

Imaging endothelial engulfment of anti-ICAM vs anti-TfR carriers. (a) Microscopy micrographs showing phase-contrast images (top panels) of ∼4.5 μm anti-ICAM vs. anti-TfR carriers after binding for 15 min at 37 °C to HUVECs, and fluorescence immunostaining (bottom panels) of NHE1 vs. clathrin heavy chain clustering at sites of carrier binding and engulfment (ring-like structures). Scale bar = 10 μm. b Quantification of the percent of bound carriers showing full ring-like engulfment structures enriched in NHE1 or clathrin heavy chain. Data are mean ± SEM, with n ≥ 40 cells. # compares anti-ICAM vs. anti-TfR carriers. ### represents p ≤ 0.001, by Students t-test
Biodistribution of anti-ICAM vs anti-TfR antibodies and antibody-coated carriers in mice
As mentioned above, effective targeting and vesicular transport are crucial elements impacting biodistribution of therapeutics. ERT for most LSDs requires efficient delivery of recombinant enzymes to peripheral organs and CNS. Targeting receptors in these tissues may enhance efficacy of therapeutic interventions.
Using radioisotope tracing, we tested antibodies and antibody-coated carriers targeted to ICAM-1 vs. TfR. As shown in Fig. 5, the circulating blood level of anti-ICAM was comparable to anti-TfR at 30 min after injection (46.9 ± 2.7 % and 42.1 ± 1.3 % of injected dose or % ID), and appreciably lower than control IgG (75.5 ± 3.7 % ID; data not shown), suggesting enhanced accumulation in tissues. We used specificity index (SI) to measure accumulation in tissues due to ICAM-1 or TfR targeting (see Methods). Both anti-ICAM and anti-TfR displayed increased uptake over control IgG in all organs, with similar accumulation in liver, an example of a RES organ enabling clearance of foreign materials (Fig. 5b). The targeting specificity for a representative peripheral organ (lungs) was higher for anti-ICAM vs. anti-TfR (Fig. 5c; 4.0-fold), while the opposite occurred in brain (Fig. 5d; 1.4-fold for TfR).

Biodistribution of anti-ICAM vs anti-TfR antibodies and antibody-coated carriers in mice. a Blood levels of 125I-labeled anti-ICAM vs. anti-TfR, or their ∼250 nm carrier counterparts measured at 30 min after i.v. injection in mice, expressed as the percentage of the injected dose (%ID). b–d Specific tissue accumulation of these formulations compared to control IgG counterparts, calculated as the specificity index (SI), which normalizes the %ID to the weight of the organ and the circulation fraction (see Methods). SI values above 1 represent specific targeting in an organ over control IgG formulations. Data are mean ± SEM, with n ≥ 3 mice. * compares antibodies vs carriers for each target, and # compares targeting to ICAM-1 vs. TfR for each formulation. * or # represent p ≤ 0.05, ** or ## represent p ≤ 0.01, and *** or ### represent p ≤ 0.001, by Students t-test
A different behavior was observed for antibody-coated carriers. Blood levels of anti-ICAM or anti-TfR carriers were considerably lower than free counterparts (Fig. 5a; ≈6.6-fold). This suggests increased removal from blood and/or organ specificity, likely due to carrier multivalency. Anti-ICAM carriers displayed increased accumulation but reduced specificity in RES organs, likely resulting from greater non-specific uptake (liver SI decrease of 2.4-fold; Fig. 5b) and enhanced accumulation and specificity in peripheral organs (1.5-fold in lungs; Fig. 5c) and brain (2.7-fold; Fig. 5d). However, specific tissue accumulation (lungs, liver, brain) decreased for anti-TfR carriers compared to free antibody counterpart (3.2-fold, 3.4-fold, 1.2-fold, respectively; Fig. 5b, c, d). Anti-TfR carriers exceeded accumulation over control IgG carriers in brain, while anti-ICAM carriers displayed specificity in both lung and brain, with even better performance than anti-TfR in brain (1.9-fold improvement). Hence, results in vivo correlate well with cell culture observations of reduced binding and endocytosis of anti-TfR carriers compared to free anti-TfR, and an opposite effect for targeting ICAM-1.
Lysosomal enzyme delivery in mice by ICAM-1- vs. TfR-targeted carriers
We finally determined the potential delivery improvement for recombinant lysosomal enzyme injected i.v. and targeted via anti-ICAM or anti-TfR carriers compared to free counterpart (as in clinical applications). For this purpose, we used recombinant ASM, an investigational ERT requiring delivery to CNS and peripheral organs for treatment of types A and B Niemann Pick disease (type A OMIM # 257200, type B OMIM # 607616) (He et al 1999).
Using similar doses of 125I-ASM, coupling to anti-ICAM or anti-TfR carriers significantly lowered blood levels of circulating enzyme by 30 min post-injection (Fig. 6a), suggesting greater removal from circulation and delivery to organs. ASM uptake was enhanced in RES and non-RES peripheral organs, and also brain for ICAM-1- and TfR-targeted carriers vs. free counterparts, with greater benefit from ICAM-1 targeting. For instance, anti-ICAM carriers enhanced specific ASM delivery compared to anti-TfR carriers by 3.0-fold in liver, 81.3-fold in lung, and 2.5-fold in brain (Fig. 6b, c, d). Therefore, targeting ICAM-1 or TfR may be valuable for lysosomal enzyme delivery, as previously shown, yet ICAM-1 targeting may offer advantages with multivalent carriers.

Lysosomal enzyme delivery in mice by ICAM-1- vs. TfR-targeted carriers. a Blood levels of 125I-acid sphingomyelinase (125I-ASM) injected i.v. in mice as a free counterpart or coupled to ∼250 nm anti-ICAM vs. anti-TfR carriers, measured at 30 min after injection and expressed as the percentage of the injected dose (%ID). b–d Specific tissue accumulation of anti-ICAM/ASM vs. anti-TfR/ASM carriers compared to free ASM, calculated as the specificity index (SI), described in Fig. 5. Data are mean ± SEM, with n ≥ 3 mice. * Compares free enzyme vs carrier-coupled enzyme for each target, and # compares targeting to ICAM-1 vs. TfR. * or # represent p ≤ 0.05, and *** or ### represent p ≤ 0.001, by Students t-test
Discussion
ERT is promising for treatment of LSDs. Yet, apart from accessible RES tissues, current delivery of lysosomal enzymes is relatively hindered to peripheral organs and, primarily, CNS (Brady 2003; Desnick and Schuchman 2002). Coupling affinity moieties targeted to receptors which enable endocytosis in a glycosylation-independent manner (either directly to lysosomal enzymes or indirectly to enzyme-loaded nanocarriers), can facilitate transport across cellular barriers (e.g., BBB) or intracellularly to lysosomes (Boado et al 2008, 2009, 2011; Chen et al 2008; Garnacho et al 2008a; Hsu et al 2012; Lebowitz et al 2004; Lu et al 2011; Muro et al 2006a; Osborn et al 2008; Prince et al 2004; Vaags et al 2005; Xia et al 2001; Zhang et al 2008; Zhou et al 2012). In this work, we have explored comparatively the binding, endocytosis, and biodistribution patterns of affinity moieties (antibodies) and antibody-coated carriers targeting ICAM-1 vs. TfR for lysosomal enzyme delivery to peripheral and CNS tissues.
Interestingly, targeting performance of ICAM-1 vs. TfR greatly depended on presentation of affinity moieties as free counterparts versus coated on polymer carriers. Free anti-ICAM and anti-TfR bound control ECs similarly, yet ICAM-1-targeted carriers displayed greater cellular targeting than TfR-targeted carriers (Fig. 1). This agreed with the biodistribution of antibodies vs. antibody-coated carriers injected intravenously in mice. For instance, antibodies targeting TfR accumulated better in brain than anti-ICAM, while accumulation of anti-ICAM carriers surpassed anti-TfR carriers in this organ (Fig. 5). This occurred despite similar carrier valency (Table 1), which should produce similarly effective targeting. Greater binding was observed for both sub-micrometer and micrometer size carriers targeting ICAM-1 vs. TfR (Figs. 1b and 2a), which was further enhanced for affinity moieties smaller than antibodies (e.g., γ3-derivative peptide vs. transferrin).
This suggests size-dependency in effectively accessing TfR on the endothelial lumen, where presentation of affinity moieties on the coat of carriers may pose steric hindrances for binding to this cell surface receptor. Such an effect may depend on the molecular location of the particular epitope targeted by the antibodies used. Anti-ICAM antibodies in our study bind to the two most membrane-distal domains on ICAM-1 (Jevnikar et al 1990; Marlin and Springer 1987). Unfortunately, this information is not available for anti-TfR antibodies used, yet inferring from homology between human and mouse TfR, the antibody used in mice may bind a membrane-distal domain of TfR (Kissel et al 1998). As an example of this concept, similar carriers directed to a membrane-proximal epitope of a related molecule (PECAM-1) lacked binding to cultured ECs vs. carriers targeted to membrane-distal epitopes, despite similar binding when presented as free counterparts (Garnacho et al 2008b). In another study the efficiency of ACE binding to endothelium in vivo varied greatly depending on the epitope targeted (Balyasnikova et al 2005).
Also related to potential steric hindrance for carrier binding, intrinsic features of the examined receptors, such as their length and location on the plasmalemma, can impact targeting. For example, ICAM-1 extends further from the endothelial lumen than TfR (∼19 nm vs ∼9 nm, respectively) (Fuchs et al 1998; Jun et al 2001) and appears to reside in luminal microvilli-like projections (Lossinsky et al 1995; Carpén et al 1992) which may be more amenable for engagement by targeted carriers. A similar effect was reported for targeting ganglioside GM1 on intestinal cells using cholera toxin B as a ligand. While FITC-labeled cholera toxin B (∼6 nm) bound cells, conjugation to particles (∼29 nm) reduced targeting, and binding was totally abolished by increasing particle size (∼1.1 μm) (Frey et al 1996).
ICAM-1 targeting with antibodies or antibody-coated carriers was superior to TfR in ECs activated with TNFα (Figs. 1 and 2). ICAM-1 is overexpressed in pathological conditions including inflammation, thrombosis, atherosclerosis, oxidative stress, and metabolic imbalance (Degraba et al 2000; Shen et al 2008; Muro 2010). Alternatively, TfR expression increases relatively modestly or responds neutrally to different inflammatory mediators (Nanami et al 2005; Visser et al 2004). Hence, selecting between these molecules for therapeutic or prophylactic interventions depends somewhat on overall and local physiological–pathological balance.
Major differences were also found regarding ICAM-1- vs. TfR-mediated vesicular uptake. Both markers support endocytosis and transcytosis (Ghaffarian et al 2012; Muro et al 2003; Pardridge and Boado 2012). However, free anti-ICAM poorly induced endocytosis, while anti-ICAM carriers were efficiently internalized by ECs, and an opposite outcome occurred for TfR targeting (Fig. 3). Indeed, anti-ICAM carriers underwent rapid engulfment by NHE1-enriched plasmalemma structures, whereas recruitment of clathrin heavy chain was less efficient for anti-TfR carriers (Fig. 4). As observed previously (Muro et al 2005), the rate of anti-ICAM carrier uptake seemed independent of initial binding level (Figs. 1b and 3b), and endocytosis led to enhanced absolute uptake of carriers compared to TfR targeting (Fig. 3c).
This highlights a different need for multivalent engagement of ICAM-1 vs. TfR, where greater receptor clustering must be achieved to induce CAM vs. clathrin-mediated endocytosis. Although the threshold required to induce endocytosis via ICAM-1 is unknown, this may reflect a different biological function. For instance, transferrin binding to TfR suffices to induce signaling cascades leading to formation of clathrin-coated pits (Conrad and Umbreit 2000; Fishman et al 1987). In contrast, multiple β2 integrins are presented on leukocytes, and “sensed” by the endothelium similarly to multivalent ICAM-1 engagement by anti-ICAM carriers. Indeed, formation of plasmalemma engulfing structures, invaginations, coalescing vesicles, upstream signaling and cytoskeletal restructuring observed during CAM-mediated endocytosis of anti-ICAM carriers are common events elicited during ICAM-1 engagement by leukocytes transmigrating across ECs (Carman and Springer 2004; Carman et al 2007; Dvorak and Feng 2001; Millán et al 2006; Barreiro et al 2008).
Therefore, it is likely that the different biodistribution patterns found in vivo for ICAM-1- vs. TfR- targeting antibodies or antibody-coated carriers are not only due to binding differences, but also to utilization of different endocytic mechanisms. Clathrin-mediated uptake and transcytosis is limited by size of natural clathrin-coated pits (∼100–150 nm, Hirst and Robinson 1998; Steven et al 1983). In contrast, ICAM-1 regulates leukocyte transmigration by a transcytosis-like mechanism without opening cell junctions, and is therefore amenable to sustained formation of large vesicular structures (Carman and Springer 2004; Dvorak and Feng 2001; Millán et al 2006). As recently reported, this apparently occurs from concerted activity of NHE1 and the sphingomyelin/ceramide pathway, which regulates plasma membrane flexibility (Hillebrand et al 2006; Serrano et al 2012). Consequently, the uptake efficiency of therapeutics and/or their carriers via TfR vs. ICAM-1 is impacted. For instance, several carrier types have been used for brain delivery via TfR (Ko et al 2009; van Rooy et al 2011; Shi et al 2001; Pang et al 2011), yet a size restriction of ∼80 nm has been reported for brain targeting of transferrin-coupled liposomes (Hatakeyama et al 2004). In contrast, endothelial ICAM-1 targeting and endocytosis has been shown in vivo even for micrometer-sized carriers (Muro et al 2008). This supports our results indicating enhanced delivery of a model lysosomal enzyme (ASM) to RES, peripheral organs and CNS by TfR- and, more prominently, ICAM-1-targeted carriers (Fig. 6).
As deduced from these observations, it is likely that smaller and less bulky fusion proteins or conjugates may benefit from targeting TfR vs. ICAM-1. Selecting between these delivery modalities depends on a variety of parameters, and both present interesting advantages and certain disadvantages. Smaller fusion proteins or conjugates provide simpler design, ready production, and facilitated diffusion in tissue parenchyma, while drug carriers can enable control of circulation time, avoid rapid degradation, limit immunological responses against recombinant enzymes, and regulate rate and location of enzyme release (Moghimi et al 2001; Muro 2010; Torchilin 2006; Matzner et al 2008; Ohashi et al 2008).
In conclusion, targeting ICAM-1 or TfR holds considerable promise to enhance delivery of lysosomal ERTs (and likely other therapeutics) to peripheral organs and CNS, where TfR likely provides greater benefit for smaller fusion proteins or conjugates, while ICAM-1 appears superior for delivery with larger multivalent carriers.
References
Altieri DC, Duperray A, Plescia J, Thornton GB, Languino LR (1995) Structural recognition of a novel fibrinogen gamma chain sequence (117–133) by intercellular adhesion molecule-1 mediates leukocyte-endothelium interaction. J Biol Chem 270(2):696–699
Balyasnikova IV, Metzger R, Visintine DJ et al (2005) Selective rat lung endothelial targeting with a new set of monoclonal antibodies to angiotensin I-converting enzyme. Pulm Pharmacol Ther 18(4):251–267
Banks WA (2009) Blood–brain barrier as a regulatory interface. Forum Nutr 63:102–110
Barreiro O, Zamai M, Yáñez-Mó M et al (2008) Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J Cell Biol 183(3):527–542
Begley DJ, Pontikis CC, Scarpa M (2008) Lysosomal storage diseases and the blood–brain barrier. Curr Pharm Des 14(16):1566–1580
Boado RJ, Zhang Y, Xia CF, Wang Y, Pardridge WM (2008) Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood brain barrier. Biotechnol Bioeng 99(2):475–484
Boado RJ, Zhang Y, Wang Y, Pardridge WM (2009) Engineering and expression of a chimeric transferrin receptor monoclonal antibody for blood–brain barrier delivery in the mouse. Biotechnol Bioeng 102(4):1251–1258
Boado RJ, Hui EK, Lu JZ, Zhou Q, Pardridge WM (2011) Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous Trojan horse-iduronidase fusion protein. Mol Pharm 8(4):1342–1350
Brady RO (2003) Enzyme replacement therapy: conception, chaos and culmination. Philos Trans R Soc Lond B Biol Sci 358(1433):915–919
Calderon AJ, Bhowmick T, Leferovich J et al (2011) Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J Control Release 150(1):37–44
Cardone M, Porto C, Tarallo A et al (2008) Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 1(1):6
Carman CV, Springer TA (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 167(2):377–388
Carman CV, Sage PT, Sciuto TE et al (2007) Transcellular diapedesis is initiated by invasive podosomes. Immunity 26(6):784–797
Carpén O, Pallai P, Staunton DE, Springer TA (1992) Association of intercellular adhesion molecule-1 (ICAM-1) with actin-containing cytoskeleton and alpha-actinin. J Cell Biol 118(5):1223–1234
Chen CH, Dellamaggiore KR, Ouellette CP et al (2008) Aptamer-based endocytosis of a lysosomal enzyme. Proc Natl Acad Sci U S A 105(41):15908–15913
Conrad ME, Umbreit JN (2000) Iron absorption and transport—an update. Am J Hematol 64(4):287–298
Dautry-Varsat A (1986) Receptor-mediated endocytosis: the intracellular journey of transferrin and its receptor. Biochimie 68(3):375–381
DeGraba T, Azhar S, Dignat-George F et al (2000) Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol 47(2):229–233
Desnick RJ, Schuchman EH (2002) Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet 3(12):954–966
Dhami R, Schuchman EH (2004) Mannose 6-phosphate receptor-mediated uptake is defective in acid sphingomyelinase-deficient macrophages: implications for Niemann-Pick disease enzyme replacement therapy. J Biol Chem 279(2):1526–1532
Dvorak AM, Feng D (2001) The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem 49(4):419–432
Fishman JB, Rubin JB, Handrahan JV, Connor JR, Fine RE (1987) Receptor-mediated transcytosis of transferrin across the blood–brain barrier. J Neurosci Res 18(2):299–304
Frey A, Giannasca KT, Weltzin R et al (1996) Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med 184(3):1045–1059
Fuchs H, Lucken U et al (1998) Structural model of phospholipid-reconstituted human transferrin receptor derived by electron microscopy. Structure 6(10):1235–1243
Futerman AH, van Meer G (2004) The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 5(7):554–565
Garnacho C, Dhami R, Simone E et al (2008a) Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther 325(2):400–408
Garnacho C, Albelda SM, Muzykantov VR, Muro S (2008b) Differential intra-endothelial delivery of polymer nanocarriers targeted to distinct PECAM-1 epitopes. J Control Release 130(3):226–233
Garnacho C, Serrano D, Muro S (2012) A fibrinogen-derived peptide provides ICAM-1-specific targeting and intra-endothelial transport of polymer nanocarriers in human cell cultures and mice. J Pharmacol Exp Ther 340(3):638–647
Ghaffarian R, Bhowmick T, Muro S (2012) Transport of nanocarriers across gastrointestinal epithelial cells by a new transcellular route induced by targeting ICAM-1. J Control Release June 12 (Epub ahead of print)
Hatakeyama H, Akita H, Maruyama K et al (2004) Factors governing the in vivo tissue uptake of transferrin-coupled polyethylene glycol liposomes in vivo. Int J Pharm 281(1–2):25–33
He X, Miranda SR, Xiong X, Dagan A, Gatt S, Schuchman EH (1999) Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim Biophys Acta 1432(2):251–264
Hillebrand U, Hausberg M, Stock C et al (2006) 17beta-estradiol increases volume, apical surface and elasticity of human endothelium mediated by Na+/H+ exchange. Cardiovasc Res 69(4):916–924
Hirst J, Robinson MS (1998) Clathrin and adaptors. Biochim Biophys Acta 1404(1–2):173–193
Hsu J, Serrano D, Bhowmick T, Muro S (2011) Enhanced endothelial delivery and biochemical effects of alpha-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release 149(3):323–331
Hsu J, Northrup L, Bhowmick T, Muro S (2012) Enhanced delivery of alpha-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine 8(5):731–739
Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KS, Mason DY (1984) Transferrin receptor on endothelium of brain capillaries. Nature 312(5990):162–163
Jevnikar AM, Wuthrich RP, Takei F et al (1990) Differing regulation and function of ICAM-1 and class II antigens on renal tubular cells. Kidney Int 38(3):417–425
Jin M, Snider MD (1993) Role of microtubules in transferrin receptor transport from the cell surface to endosomes and the Golgi complex. J Biol Chem 268(24):18390–18397
Jun CD, Carman CV, Redick SD, Shimaoka M, Erickson HP, Springer TA (2001) Ultrastructure and function of dimeric, soluble intercellular adhesion molecule-1 (ICAM-1). J Biol Chem 276(31):29019–29027
Kissel K, Hamm S et al (1998) Immunohistochemical localization of the murine transferrin receptor (TfR) on blood–tissue barriers using a novel anti-TfR monoclonal antibody. Histochem Cell Biol 110(1):63–72
Ko YT, Bhattacharya R, Bickel U (2009) Liposome encapsulated polyethylenimine/ODN polyplexes for brain targeting. J Control Release 133(3):230–237
LeBowitz JH, Grubb JH, Maga JA, Schmiel DH, Vogler C, Sly WS (2004) Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc Natl Acad Sci U S A 101(9):3083–3088
Lossinsky AS, Mossakowski MJ, Pluta R, Wisniewski HM (1995) Intercellular adhesion molecule-1 (ICAM-1) upregulation in human brain tumors as an expression of increased blood–brain barrier permeability. Brain Pathol 5(4):339–344
Lu JZ, Boado RJ, Hui EK, Zhou QH, Pardridge WM (2011) Expression in CHO cells and pharmacokinetics and brain uptake in the Rhesus monkey of an IgG-iduronate-2-sulfatase fusion protein. Biotechnol Bioeng 108(8):1954–1964
Marlin SD, Springer TA (1987) Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell 51(5):813–819
Matzner U, Matthes F, Weigelt C et al (2008) Non-inhibitory antibodies impede lysosomal storage reduction during enzyme replacement therapy of a lysosomal storage disease. J Mol Med 86(4):433–442
Millán J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ (2006) Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol 8(2):113–123
Mistry PK, Wraight EP, Cox TM (1996) Therapeutic delivery of proteins to macrophages: implications for treatment of Gaucher’s disease. Lancet 348(9041):1555–1559
Moghimi SM, Hunter AC, Murray JC (2001) Long circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 53(2):283–318
Murciano JC, Muro S, Koniaris L et al (2003) ICAM-directed vascular immunotargeting of antithrombotic agents to the endothelial luminal surface. Blood 101(10):3977–3984
Muro S (2010) New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2(2):189–204
Muro S, Wiewrodt R, Thomas A et al (2003) A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci 116(Pt 8):1599–1609
Muro S, Gajewski C, Koval M, Muzykantov VR (2005) ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood 105(2):650–658
Muro S, Schuchman EH, Muzykantov VR (2006a) Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther 13(1):135–141
Muro S, Mateescu M, Gajewski C et al (2006b) Control of intracellular trafficking of ICAM-1-targeted nanocarriers by endothelial Na+/H+ exchanger proteins. Am J Physiol Lung Cell Mol Physiol 290(5):L809–L817
Muro S, Garnacho C, Champion JA et al (2008) Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther 16(8):1450–1458
Nanami M, Ookawara T, Otaki Y et al (2005) Tumor necrosis factor-alpha-induced iron sequestration and oxidative stress in human endothelial cells. Arterioscler Thromb Vasc Biol 25(12):2495–2501
Ohashi T, Iizuka S, Ida H, Eto Y (2008) Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. Mol Genet Metab 94(3):313–318
Osborn MJ, McElmurry RT, Peacock B, Tolar J, Blazar BR (2008) Targeting of the CNS in MPS-IH using anonviral transferrin-alpha-L-iduronidase fusion gene product. Mol Ther 16(8):1459–1466
Pang Z, Gao H, Yu Y et al (2011) Brain delivery and cellular internalization mechanisms for transferrin conjugated biodegradable polymersomes. Int J Pharm 415(1–2):284–292
Pardridge WM (2010) Biopharmaceutical drug targeting to the brain. J Drug Target 18(3):157–167
Pardridge WM, Boado RJ (2012) Reengineering biopharmaceuticals for targeted delivery across the blood–brain barrier. Methods Enzymol 503:269–292
Prince WS, McCormick LM, Wendt DJ et al (2004) Lipoprotein receptor binding, cellular uptake, and lysosomal delivery of fusions between the receptor-associated protein (RAP) and alpha-L-iduronidase or acid alpha-glucosidase. J Biol Chem 279(33):35037–35046
Rothlein R, Dustin ML, Marlin SD, Springer TA (1986) A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol 137(4):1270–1274
Schnitzer JE (2001) Caveolae: from basic trafficking mechanisms to targeting transcytosis for tissue-specific drug and gene delivery in vivo. Adv Drug Deliv Rev 49(3):265–280
Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S (2012) Intercellular adhesion molecule 1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler Thromb Vasc Biol 32(5):1178–1185
Shen JS, Meng XL, Moore DF, Quirk JM, Shayman JA, Schiffmann R, Kaneski CR (2008) Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab 95(3):163–168
Shi N, Boado RJ, Pardridge WM (2001) Receptor-mediated gene targeting to tissues in vivo following intravenous administration of pegylated immunoliposomes. Pharm Res 18(8):1091–1095
Steven AC, Hainfeld JF, Wall JS, Steer CJ (1983) Mass distributions of coated vesicles isolated from liver and brain: analysis by scanning transmission electron microscopy. J Cell Biol 97(6):1714–1723
Torchilin VP (2006) Multifunctional nanocarriers. Adv Drug Deliv Rev 58(14):1532–1555
Urayama A, Grubb JH, Sly WS, Banks WA (2004) Developmentally regulated mannose 6-phosphate receptor-mediated transport of a lysosomal enzyme across the blood–brain barrier. Proc Natl Acad Sci U S A 101(34):12658–12663
Vaags AK, Campbell TN, Choy FY (2005) HIV TAT variants differentially influence the production of glucocerebrosidase in Sf9 cells. Genet Mol Res 4(3):491–495
van Rooy I, Mastrobattista E, Storm G, Hennink WE, Schiffelers RM (2011) Comparison of five different targeting ligands to enhance accumulation of liposomes into the brain. J Control Release 150(1):30–36
Visser CC, Voorwinden LH, Crommelin DJ, Danhof M, de Boer AG (2004) Characterization and modulation of the transferrin receptor on brain capillary endothelial cells. Pharm Res 21(5):761–769
Xia H, Anderson B, Mao Q, Davidson BL (2000) Recombinant human adenovirus: targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J Virol 74(23):11359–11366
Xia H, Mao Q, Davidson BL (2001) The HIV Tat protein transduction domain improves the biodistribution of beta-glucuronidase expressed from recombinant viral vectors. Nat Biotechnol 19(7):640–644
Zhang XY, Dinh A, Cronin J, Li SC, Reiser J (2008) Cellular uptake and lysosomal delivery of galactocerebrosidase tagged with the HIV Tat protein transduction domain. J Neurochem 104(4):1055–1064
Zhou QH, Boado RJ, Lu JZ, Hui EK, Pardridge WM (2012) Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab Dispos 40(2):329–335
Acknowledgments
This study was funded by grants from the American Heart Association (09BGIA2450014) and National Institutes of Health (R01 HL098416) to SM.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Frits Wijburg
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Fluorescence microscopy of antibody binding to ICAM-1 vs. TfR on endothelial cells. Control or TNFα-activated HUVECs were incubated with anti-ICAM or anti-TfR for 15 min at 37 °C. Cells were washed to remove unbound antibody, fixed, and stained with FITC-conjugated goat anti-mouse IgG. Phase-contrast images were used to delimit cell borders (dashed lines). Scale bar = 10 μm. (JPEG 43 kb)
Supplementary Fig. 2
Fluorescence microscopy of carrier binding to ICAM-1 vs. TfR on endothelial cells. Binding of ∼250 nm FITC-labeled anti-ICAM vs. anti-TfR carriers to control or TNFα-activated HUVECs after 1 h incubation. Cells were subsequently washed to remove unbound carriers. Phase-contrast images were used to delimit cell borders (dashed lines). Scale bar = 10 μm. (JPEG 36 kb)
Supplementary Fig. 3
Fluorescence microscopy of endocytosis of antibodies targeted to endothelial ICAM-1 vs. TfR. Uptake of anti-ICAM vs. anti-TfR by TNFα-activated HUVECs, assessed after 1 h incubation at 37 °C. Unbound antibodies were washed off and surface-bound antibodies were stained with a Texas-Red secondary IgG. Cells were then permeabilized and antibodies located both at the surface + internalized were stained with FITC-labeled secondary IgG. Internalized antibodies appear as FITC single-labeled in green, while surface-bound antibodies display FITC + Texas-Red double-labeled yellow color. Phase-contrast images were used to delimit cell borders (dashed lines). Scale bar = 10 μm. (JPEG 23 kb)
Supplementary Fig. 4
Fluorescence microscopy of endocytosis of carriers targeted to endothelial ICAM-1 vs. TfR. Uptake of ∼250 nm FITC-labeled anti-ICAM vs. anti-TfR carriers, assessed after 1 h incubation at 37 °C with control or TNFα-activated HUVECs. Unbound carriers were removed and surface-bound carriers were stained with a Texas-Red secondary IgG. Internalized carriers appear as FITC single-labeled in green, while surface-bound carriers display FITC + Texas-Red double-labeled yellow color. Phase-contrast images were used to delimit cell borders (dashed lines). Scale bar = 10 μm. (JPEG 53 kb)
Rights and permissions
About this article
Cite this article
Papademetriou, J., Garnacho, C., Serrano, D. et al. Comparative binding, endocytosis, and biodistribution of antibodies and antibody-coated carriers for targeted delivery of lysosomal enzymes to ICAM-1 versus transferrin receptor. J Inherit Metab Dis 36, 467–477 (2013). https://doi.org/10.1007/s10545-012-9534-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-012-9534-6