
Abstract
Recessive mutations in the mitochondrial arginyl-transfer RNA synthetase (RARS2) gene have been associated with early onset encephalopathy with signs of oxidative phosphorylation defects classified as pontocerebellar hypoplasia 6. We describe clinical, neuroimaging and molecular features on five patients from three unrelated families who displayed mutations in RARS2. All patients rapidly developed a neonatal or early-infantile epileptic encephalopathy with intractable seizures. The long-term follow-up revealed a virtual absence of psychomotor development, progressive microcephaly, and feeding difficulties. Mitochondrial respiratory chain enzymes in muscle and fibroblasts were normal in two. Blood and CSF lactate was abnormally elevated in all five patients at early stages while appearing only occasionally abnormal with the progression of the disease. Cerebellar vermis hypoplasia with normal aspect of the cerebral and cerebellar hemispheres appeared within the first months of life at brain MRI. In three patients follow-up neuroimaging revealed a progressive pontocerebellar and cerebral cortical atrophy. Molecular investigations of RARS2 disclosed the c.25A>G/p.I9V and the c.1586+3A>T in family A, the c.734G>A/p.R245Q and the c.1406G>A/p.R469H in family B, and the c.721T>A/p.W241R and c.35A>G/p.Q12R in family C. Functional complementation studies in Saccharomyces cerevisiae showed that mutation MSR1-R531H (equivalent to human p.R469H) abolished respiration whereas the MSR1-R306Q strain (corresponding to p.R245Q) displayed a reduced growth on non-fermentable YPG medium. Although mutations functionally disrupted yeast we found a relatively well preserved arginine aminoacylation of mitochondrial tRNA. Clinical and neuroimaging findings are important clues to raise suspicion and to reach diagnostic accuracy for RARS2 mutations considering that biochemical abnormalities may be absent in muscle biopsy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The main role of mitochondria is to convert energy derived from oxidation of substrates into the high-energy bond of ATP by the process of oxidative phosphorylation (OXPHOS). The elevated dependence for sufficient ATP production in the developing central nervous system (CNS) easily explains the high frequency of mitochondrial encephalopathies in infancy if the pathway is hampered. However, the relatively limited spectrum of clinical presentations observed at this age is challenged by an ever growing number of genetic mutations affecting not only the many components of the OXPHOS system but also the numerous subsidiary proteins required for the correct functioning of the mitochondrial proteome and the complex interaction between the two genomes controlling oxidative metabolism (DiMauro 2011). In the face of these complexities and the many possible etiologies, reaching a conclusive diagnosis is hard and laborious, and the molecular defect causing different syndromes often remains unexplained (McFarland et al 2010; Wong 2010). Thus, systematic clinical descriptions of relatively rarer conditions, as well as identification of neuroimaging clues or sensitive biomarkers are of utmost relevance when searching the etiology of childhood-onset encephalopathies.
Three of five siblings in a Sephardic Jewish kindred were reported in 2007 with an infantile encephalopathy resembling pontocerebellar hypoplasia (PCH) (Edvardson et al 2007). Early MRI showed cerebellar and vermian hypoplasia with normal brain volume in one patient, whereas follow-up scans revealed progressive atrophy of the supra- and subtentorial brain structures, and white matter depletion in two other sibs (Edvardson et al 2007). Multiple, though variable, respiratory chain enzyme (RC) defects in muscle and elevated blood and CSF lactate levels were observed and a clinical diagnosis of PCH6 was entertained. A homozygous splice-site mutation in RARS2, the gene encoding mitochondrial arginyl-transfer RNA synthetase (mt-ArgRS), was ultimately revealed. Following the original description, three further patients harboring mutations in RARS2 have been reported (Rankin et al 2010; Namavar et al 2011; Glamuzina et al 2011) fitting the clinical diagnosis of PCH6. An additional case has been described presenting also anterior horn cell involvement, as seen in PCH1 (Namavar et al 2011).
Accurate translation of mRNA into protein is a fundamental step for maintenance of cellular integrity. Translational fidelity is achieved by two key events: synthesis of correctly paired aminoacyl-tRNAs by aminoacyl-tRNA synthetases (ARSs) and stringent selection of aminoacyl-tRNAs by the ribosome. ARSs are ubiquitous enzymes especially needed for attaching amino acids to their cognate tRNA molecules in the cytoplasm and mitochondria (Ling et al 2009). Thus, it is not too surprising that mutations in genes encoding ARSs are increasingly being recognized as causes of human pathologies, frequently affecting the brain (Antonellis and Green 2008; Chrzanowska-Lightowlers et al 2011).
We describe the clinical and molecular features of five patients from three unrelated families who displayed mutations in RARS2. All the patients were affected by an early-onset, rapidly progressive cerebral and ponto-cerebellar atrophy with intractable epilepsy. We detected new mutations and studied their functional properties using “budding” yeast as a model organism for cellular respiration insufficiency. We also emphasize the clinical and neuroimaging features which should raise early suspicion for defective RARS2.
Materials and methods
Patients
Patients were ascertained at the Department of Neurosciences of the Bambino Gesù Hospital. Their follow up has lasted over 10 years and included repeated neurological, electrophysiological, and neuroimaging examinations.
Molecular genetics
Total DNA was extracted from tissues according to standard salting-out procedures. The coding exons and exon–intron boundaries of RARS2 were PCR-amplified, and amplified fragments were purified and bidirectionally analyzed using capillary Sanger sequencing. To confirm the mutations identified, rule out their presence in 300 control chromosomes, and assess parent-of-origin, specific primer extension techniques were employed using minisequencing analysis (ABI Prism SNaPshot minisequencing kit, Applied Biosystems, Foster City, CA). To search a posteriori for allelic sharing between the two sibling in families A and B, a total of 85 microsatellite markers included in the ABI PRISM Linkage Mapping Set v2.5-MD5 (Applied Biosystems, Foster City, CA, USA) and located around the selected genes causing PCH (TSEN54, TSEN34, TSEN15, and TSEN2), were genotyped as described elsewhere (Cassandrini et al 2010). To test the effects on mRNA splicing of the mutations identified in cases A-01 and C-01, total cellular RNA was extracted from cultured fibroblasts using a microscale total RNA separator kit (Ambion INC., Austin, TX). Four overlapping fragments encompassing the entire coding region of RARS2 were directly amplified from the patient’s total RNA with the Superscript First-Strand Synthesis System kit (Life Technology, Gaithersburg, MD). PCR products of relevant fragments were cloned into the PCR cloning vector pCR 2.1-TOPO (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. After transformation in TOPO bacteria, the clones were purified and sequenced in both directions.
Real-time quantitative PCR (q-PCR)
Messenger RNA/RARS2 transcript level was determined in patients’ cultured skin fibroblasts. For q-PCR runs in an ABI7500Fast (Applied Biosystems), we used the TaqMan Universal PCR Protocol, human RARS2 (Hs00368084_m1, Applied Biosystems) as probe, GAPDH (Hs99999905_m1, Applied Biosystems) for endogenous normalization, and the comparative Ct method (Livak and Schmittgen 2001).
Western blotting
In cultured skin fibroblast mitochondria, immunolabeling used an anti-mouse polyclonal RARS2 antibody (Abnova, Corporation, Taipei, Taiwan) (1:300) and a monoclonal antibody (VDAC/porin, Mitoscience, Eugene, OR) (1:12000), as a control for protein loading. Reactive bands were detected using the Immobilon Western Chemiluminescent HRP Substrate detection kit (Millipore Corporation, Billerica, MA, USA) and densitometrically quantified using Quantity One software (BioRad, Hercules, USA). Each sample was run in triplicate and normalized values were averaged and compared to normal values. Controls were age-matched children who had undergone a diagnostic punch skin biopsy and were ultimately deemed free of a defect of oxidative metabolism.
Arginyl-tRNA synthetase enzyme activity
The activity of mt-ArgRS (EC 6.1.1.19) was assayed in mitochondria purified from cultured skin fibroblasts. The mt-ArgRS enzyme activity was measured by a modification of the method previously described (Chang et al 1983, 1984). Briefly, 100 μg of mitochondrial proteins were incubated in 50 mM Tris–HCl buffer (pH 7.5), 1.25 mM MgAc2, 1.2 mM ATP, and 6 mM L-arginine. Samples (0.5 μl) were incubated at 37°C. The reaction was started by adding mitochondria and stopped by adding 0.25 ml 10 % trichloricacetic acid. For colorimetric assay, the reaction mixture was centrifuged at 2000 g for 10 min and 250 μl of the supernatant was monitored for releasing of Pi (Naito 1975; Lloyd et al 1995). All chemicals were highly purified and from Sigma-Aldrich (MO, USA).
Analysis of stability and aminoacylation of mitochondrial tRNA for arginine
Total skin fibroblast RNA was extracted under acidic conditions to preserve aminoacylated tRNAs as described (Enriquez and Attardi 1996) with minor modifications. To investigate the effect of the mutations on tRNA stability, we determined mt-tRNA for arginine (mt-tRNAArg ) steady state levels, as described previously using mt-tRNA for leucine (mt-tRNALeu) as endogenous control for loading (Tuppen et al 2008). The analysis of aminoacylation level in vivo was performed by hybridization with the same probes after separation of 3 μg of RNA on 8 % non-denaturing polyacrylamide (29:1) gel in 0.1 M sodium acetate (pH 5.0) buffer. To deacylate tRNAs, an aliquot of total RNA was boiled for 10 min (pH 8.0) before electrophoresis.
Construction and analysis of yeast models and mutant RARS2
The human mutations were modeled on the orthologous yeast MSR1 gene. A single MSR1 allele in a wild type, diploid W303 strain was inactivated by homologous recombination with a HIS3 cassette. The resulting cells were then transformed with a centromeric plasmid containing either the wild type or one of the mutant MSR1 alleles and a URA3 selection marker. Two different set of plasmids were used: standard pCM189, in which expression of the gene of interest is driven by the CYC1 promoter, and a modified pCM189 where the CYC1 promoter was replaced by the endogenous MSR1 promoter. Heterozygous diploid cells were induced to sporulate and generate haploid yeast carrying a deletion of chromosomal MSR1, and different MSR1 alleles (either wild-type or individual mutants) on the episomal plasmid. Each tetrad was mechanically dissected to isolate haploid daughter cells (see for details Supplementary method 1; Supplementary Fig. 1).
Expression of rars in zebrafish
The human sequence of RARS2 was blasted to the zebrafish genome using the tblastn tool on NCBI (http://blast.ncbi.nlm.nih.gov/). Amino acid sequence was analyzed with clustalW on EBI (http://www.ebi.ac.uk/) and with PROSITE on Expasy (http://www.expasy.ch/) to identify functional domains conserved during evolution. Purification of total RNA from 5–10 embryos at different stages of development, reverse transcription, conditions for mRNA expression of zf-rars2 by qPCR, and whole-mount in situ hybridization (WMISH) (Thisse and Thisse 2008) are described in Supplementary method 2.
Statistical analyses
Student's t-test was performed with the Statistica 7 software package (Statsoft ©, Tulsa, OK). Only p values of less than 0.01 were considered significant.
Results
Patients
Between 2008 and 2010 we prospectively and consecutively collected three unrelated Italian families, five children (three boys and two girls) who were clinically suspected of RARS2 mutations and were followed from the beginning of symptoms at one single reference center. All patients manifested comparable symptoms of a severe epileptic encephalopathy since the neonatal period. Parents in all three families were healthy and denied consanguinity. In all the patients repeated cardiac, ocular, liver, and renal evaluations yielded normal findings, whereas the follow-up was complicated by a severe disabling dystonic posturing and early-onset gastroesophageal reflux which was demonstrated by esophageal pH monitoring. Severe progressive feeding difficulties required gastrostomy. Small for gestational age (SGA) was defined as live-born infants with weight below the 10th percentile for its gestational age (Peleg et al 1998). Gestational age was based on a reliable, self-reported estimate of last menstrual period or an ultrasound early in pregnancy.
The index patient (A-01) of the first family (Fig. 1), was born at term, SGA with head circumference in the 25th percentile. He presented the first day of life with stupor and persistent severe lactic acidosis in urines and blood, and marked irritability and hypotonia. Seizures started at 11 days of life and were polymorphic with apneic spells and focal clonic seizures involving alternately both sides of the body. The first brain MRI performed at 1 month of age showed cerebellar vermis hypoplasia and mild cortical atrophy (Fig. 2a, b). At 1 year, head circumference dropped below the 25th percentile. The child showed daily intractable seizures despite antiepileptic drug (AED) polytherapy. A skeletal muscle biopsy was morphologically unremarkable. Enzyme activities of mitochondrial RC in muscle homogenate showed reduced complexes I and IV with residual activities being 49 % and 55 % of the lowest control values upon correction for the levels of citrate synthase (CS). These defects were confirmed in cultured skin fibroblasts. At 2 years, the clinical condition had remained severe whereas lactic acidosis was no longer detected in blood and urine. Worsening of cortical and cerebellar atrophy was seen in serial MRI scans performed at two and three years of age paralleling the progressive microcephaly. Qualitative and quantitative alterations in the mitochondrial genome as well as point mutations in the whole mtDNA were ruled out in muscle. During the 11-year follow-up, case A-01 developed spastic quadriplegia with severe developmental delay, and even more aggressive intractable epilepsy. The younger brother (patient A-02), born at term, was also SGA and his head circumference was in the 10th percentile. Since the first days of life, he was hypotonic and poorly responsive. At three months of life, he presented with seizures which rapidly became intractable. Increased lactate was found in blood (4.5 mM/L) and the CSF (4.44 mM/L, normal <2.22 mM/L), while organic acids were not detected in urine. Brain MRI showed mild cerebellar vermis hypoplasia and moderate brain atrophy. Alike his brother, gastroesophageal reflux was diagnosed at age 6 months. RC enzyme activities in muscle and skin fibroblasts showed biochemical defects similar to his eldest brother (Table 1). The 9-year follow-up confirmed a severe psychomotor delay with progressive microcephaly, spastic quadriplegia and intractable epilepsy. Follow-up neuroimaging at ages 4 months and 3 years revealed progressive cerebellar and cerebral atrophy.

Pedigrees of the five patients from three Italian families harboring mutations in RARS2

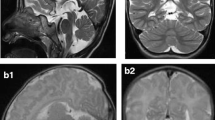
Coronal and sagittal T1 weighted images corresponding to patients A-01 (a, b) and B-02 (c), performed within the age of 1 month exemplifying the range of neuroimage severity of early cerebellar involvement from clear cerebellar hypoplasia/atrophy (a, b) to normal aspect (c). Brain MR-S of patient B-02 (d). ROI was placed at the frontal lobe and basal ganglia and the spectrum shown was obtained from frontal lobe showing a clear lactate peak (arrow)
The index patient in family B (patient B-01) was born at term with a weight appropriate for gestational age after an uneventful pregnancy. Her head circumference was in the 5th percentile. Soon after birth, she presented with unexplained repeated episodes of hypoglycemia. At the age of 20 days, she developed seizures progressing into epileptic status. In the first months of life, lactate was repeatedly found increased both in blood and CSF (2–3 times normal) (Table 1). Brain MRI performed at 4 months showed moderate diffuse brain atrophy. MRI spectroscopy (MR-S) revealed an increased lactate peak. RC activities in muscle homogenate showed normal results. At 1 year, the patient showed a severe neurodevelopmental delay and intractable epilepsy. Head circumference dropped below the 3rd percentile, and MRI and MR-S confirmed a progressive cerebral, cerebellar and pontine atrophy (Figs. 3a–c and 4a–d) as well as lactic peak. Gastrostomy was performed at age 5 years for severe gastroesophageal reflux. During the 9-year follow-up the patient continued to suffer from intractable multifocal seizures without overt improvement. Patient B-02, the younger sister of case B-01, was born at term SGA after a normal pregnancy. Head circumference was in the 20th percentile. She presented at 11 days of life with focal clonic seizures involving alternately both sides of the body. Brain MRI at 26 days was normal (Fig. 2c) but MR-S resolved a lactate peak (Fig. 2d). Mild lactic acidemia was detected. At age 2 years, head circumference had dropped below the 3rd percentile, and MRI showed severe cortical and ponto-cerebellar atrophy. At 3 years of age she continues to suffer from intractable epilepsy and severe psychomotor delay.

Sagittal T1 weighted serial neuroimages of patients B-01 (a-c) and C-01 (d-f) performed in each patient at age 4 months (a, d), 1-year (b, e), and 2-years (c, f) showing atrophy of the cerebrum, cerebellum and brainstem

Serial T2 axial neuroimages of patient B-01 perfomed at ages 2 months (a), 4 months (b), 12 months (c), 4-years (d) showing progressive progressive white matter and cortical volume loss
Patient C-01 is a 4-year-old boy who was born after an unremarkable pregnancy from urelated parents. His head circumference at birth was on the 10th percentile and birth weight was 2900 at 40 weeks. The patient presented at 20 days of life with severe hypotonia and intractable seizures. Lactate was markedly elevated in both blood and urine (three-fold normal values) but not in the CSF (Table 1). At age 4 months, brain MRI showed marked cerebellar hypoplasia and atrophy (Fig. 3d) and MR-S revealed an elevated lactate peak. There were neither histological nor biochemical alterations of oxidative metabolism in muscle and cultured skin fibroblasts at age 5 months. A severe gastroesophageal reflux was diagnosed at 2 months of age. At 1 year, head circumference dropped below the 3rd percentile. Lactic acidosis was no longer consistently detected in blood and urine. The patient continues to suffer from intractable multifocal seizures without improvement. Serial MRI scans performed at the ages of 12 months, and 2 years confirmed the progression to severe cortical and ponto-cerebellar atrophy (Fig. 3e–f).
Clinical features of the five patients are summarized in Table 1 and are compared with other reported patients with mutations in RARS2. From these findings the disease appears to affect selectively the brain with first symptoms appearing in the neonatal period and manifesting with severe hypotonia, intractable seizures, and lactic acidosis. A highly similar EEG pattern was seen in all five patients and consisted of diffuse slowing and bilateral, multifocal epileptiform abnormalities at onset with progressive deterioration and no response to poly-AED therapy. After the first year of life, all children showed very frequent myoclonic jerks in upper limbs.
Cerebellar vermis hypoplasia with normal aspect of the cerebral and cerebellar hemispheres appeared within the first months of life (Fig. 2a). In three patients follow-up scans revealed progressive cortical and ponto-cerebellar atrophy. MR-S in three patients demonstrated a lactate peak. Blood and CSF lactate levels were also elevated at early stages, but only occasionally as the disease progressed.
Molecular genetics
Qualitative and quantitative changes in the coding exons of genes associated with more common forms of PCH — namely, subunits of the TSEN complex (Budde et al 2008) and VRK1 (Renbaum et al 2009) —, progressive cerebellocerebral atrophy (PCCA) ( SepSecS ; Agamy et al 2010) or severe mitochondrial progressive encephalopathy of infancy (i.e., POLG and PEO1/Twinkle) had been ruled out in all patients at some stage in the disease course. Mutations in the coding exons of CLN1, CLN2, and CLN10 had also been excluded.
Molecular investigations of RARS2 disclosed potentially pathogenic mutations in all the children (Supplementary Fig. 2A, B). Patients A-01 and A-02 harbored the paternal c.25A>G/p.I9V in compound heterozygosity with the maternal c.1586+3A>T. The p.I9V mutation is located in the sequence of the mitochondrial leader peptide but prediction on probability to export to mitochondria (http://ihg2.helmholtz-muenchen.de/ihg/mitoprot.html; gpcr.biocomp.unibo.it/bacello/pred.htm) suggested a modest effect (0.781 wild-type; 0.736 mutant). Although the mutation was absent in a large set of controls and not reported in SNP databases (www.ncbi.nlm.nih.gov/ SNP; www.1000genomes.org), Polyphen2 prediction (http://genetics.bwh.harvard.edu/pph2) suggested a weak impact on protein structure. In cultured skin fibroblasts, the c.25A>G/p.I9V was expressed (not shown) whereas the c.1586+3A>T change produced skipping of exon 18 (Supplementary Fig. 2B), deleted 25 residues (R504_L528) in the expected anticodon-binding domain of mt-ArgRS (DALR domain), re-establishing the frame after residue S529, and predicted a slightly shortened but functional version of the protein. Patients B-01 and B-02 displayed the c.734G>A/p.R245Q on the maternal allele and the c.1406G>A/p.R469H on the paternal chromosomes. Whilst residue R245 is placed in the core region of mt-ArgRS, R469 also resides in the DALR domain. According to SIFT (http://blocks.fhcrc.org/sift/SIFT.html) and Polyphen2 prediction tools, both missense mutations were probably damaging. Patient C-01 had inherited from his father the c.721T>A/p.W241R mutation in the core region of mt-ArgRS and from his mother the already reported c.35A>G/p.Q12R (Rankin et al 2010; Namavar et al 2011). It has already been demonstrated that p.Q12R interferes with a splicing-enhancer element. In cDNA from cultured skin fibroblasts from case C-01, p.Q12R produced abnormal splicing with retention of 221-bp of intron 1, consequent frameshift, and protein truncation expected at residue 25 (p.Q12fsX25). The p.W241R mutation is predicted to be probably damaging upon SIFT, Polyphen2, and PMut (http://mmb.pcb.ub.es/PMut/PMut.jsp) analyses. None of the mutations identified in this study were detected in 300 healthy, ethnically-matched control chromosomes by mini-sequencing (Cassandrini et al 2006). A posteriori haplotype reconstruction in patients from families A and B showed concordance for polymorphic markers flanking RARS2 but neither for known PCH genes nor SepSecS.
Messenger RNA/RARS2 expression was in the low-normal range of controls when measured by q-PCR in cultured skin fibroblasts from A-01, B-02 and C-01 (Fig. 5a, upper panel). A reduction in the amount of mt-tRNAArg was found in fibroblasts from A-01, B-02 and C-01 (35, 22, and 20 % of control mean, respectively) and the residual tRNAArg transcript was almost fully acylated suggesting that uncharged mitochondrial transcripts become unstable. Mt-ArgRS enzyme activity was 2.548±0.39 in controls (n = 6) upon correction for CS activity whereas it was severely reduced in patients B-02 and C-01 (residual activities were 33 % and 19 % of control mean, respectively). The defect was less profound in case A-01 (68 % of control mean). Western blotting in skin fibroblasts mitochondria from patients A-01, B-02 and C-01 showed that the level of the protein was of correct size but reduced to 79 %, 35 %, and 28 %, respectively, of the control mean value after normalization to the immunodetection of VDAC/porin (Fig. 5b, lower panel).

Messenger RNA and protein expression in cultured skin fibroblasts from patients harboring mutations in RARS2 as compared to healthy controls. (a) Total cellular mRNA expression levels expressed as percentage of control mean. Values were obtained by qPCR (see text for details). (b) (left) Expression levels of the RARS2 gene product calculated as an arbitrary index upon correction for the levels of porin/VDAC. (right) A representative Western blotting is shown
To validate in vivo the pathogenic role of selected novel RARS2 mutations, we performed functional complementation studies using the facultative aerobic yeast Saccharomyces cerevisiae. As seen in Fig. 6, all mutants were able to grow in YPD medium containing a fermentable sugar. Mutation MSR1-R531H (equivalent to the human p.R469H) completely abolished respiration, and strains harboring the MSR1-R306Q mutation (equivalent to human p.R245Q) displayed a reduced growth on non-fermentable YPG medium. By contrast, there was practically no difference between the growth characteristics of the wild type strain and MSR1-W302R allele (homologous to p.W241R). Tetracycline inclusion in the growth medium — which represses expression from pCM189 plasmids — did not affect the growth profile of the latter two alleles (not shown). We did not model the p.I9V variant in MSR1 because isoleucine 9 is not conserved in yeast.

Growth of ΔMSR1 yeast cells harboring different MSR1 alleles on a centromeric episomal pCM189 plasmid. (a) Cells were grown in agar plates with fermentable YPD medium (left) or non-fermentable YPG medium (right) for four days. The initial suspension at 1 unit of A600 nm/mL was diluted 1/10 four times. Growth on fermentable substrates is preserved in all strains, while respiratory growth is impaired in the p.R306Q and p.R531H mutants. (b) The experiment was repeated with the p.W302R mutant using a modified pCM189 vector in which expression is driven by the physiological MSR1 promoter. Respiratory growth is maintained even with these experimental conditions. The mutant transformed with the empty vector displays reduced growth also in fermentable substrates because of the petite phenotype
Whole-mount studies in zebrafish showed that zf-rars2 mRNA is expressed ubiquitously with a stronger signal within the head and brain at 24hpf, when organogenesis is complete. Supplementary Fig. 3 shows pattern of spatio-temporal expression of zf-rars2 .
Discussion
Infantile-onset diseases due to mutations in nuclear genes encoding proteins targeted to the mitochondria are more common than primary mutations in the mitochondrial genome itself and their number increases at a rapid pace (McFarland and Turnbull 2009; Nogueira et al 2011; Chrzanowska-Lightowlers et al 2011). A mutation in any of these has the likelihood to produce severe biochemical defects of OXPHOS or be harmful for mtDNA maintenance, or both. As a consequence, clinical presentations may vary but patients — especially infants and children — frequently suffer from severe neurological illness because the developing brain has the highest request in oxidative substrates for functioning.
A similar pattern of early-onset epileptic encephalopathy and progressive cerebello-cerebral encephalopathy are common to our patients and represent useful clues for screening RARS2 particularly if associated with early lactic acidosis and lactate peak at MR-spectroscopy. Early onset seizures have also been observed in previously reported RARS2 patients (Edvardson et al 2007; Rankin et al 2010; Namavar et al 2011; Glamuzina et al 2011) but lactic acidosis may be overlooked as disease progresses and its absence should not preclude RARS2 testing. This is important for very early diagnosis considering that in individual patients a typical neuroimaging pattern of PCH may be missed. At the long-term follow-up, all patients showed progressive microcephaly, spastic quadriplegia, and virtual absence of psychomotor development. Gastroesophageal reflux was also particularly severe in our cases as disease progressed. A high incidence of feeding problems in children with mitochondrial disorders has been well established (Hom and Lavine 2004), although it remains a non specific association. Considering that the first signs of encephalopathy such as diffuse hypotonia and poor eye contact were already present in the neonatal period, that three patients in our study were SGA, and three cases (A-02, B-01 and C-01) also had a relative microcephaly at birth, it seems obvious that the genetic condition has a prenatal onset in most patients and affected mostly the brain.
Even more important hints for a correct diagnosis derived by MR-images disclosing vermis cerebellar hypoplasia during the first months of life in all the patients but one case. In three patients, follow up MRI revealed ensuing cerebral cortical atrophy together with progressive pontocerebellar atrophy and confirmed described aspects of white matter “depletion” (Edvardson et al 2007). Interestingly, basal ganglia involvement was never detected in neuroimaging studies, a quite peculiar finding for a metabolic encephalopathy related to defective OXPHOS (Mitochondrial Medicine Society's Committee on Diagnosis et al 2008; Friedman et al 2010).
Given the clinical and characteristic neuroimaging pattern related to mutations in RARS2 the differential diagnosis with other neurodegenerative conditions starting in early infancy such as neuronal ceroid-lipofuscinoses and Alpers-Huttenlocher syndrome is not complex. Another condition to be considered is PCCA, in non-dysmorphic patients (Ben-Zeev et al 2003). In the latter case, however, the degree of clinical features and absence of lactate peaks are sufficiently distinctive to pose a correct diagnosis besides the analysis of the SepSecS gene (Agamy et al 2010).
In this work we identified independent mutations of RARS2 in unrelated individuals presenting with exquisitely similar phenotypes. A firm genetic diagnosis was supported by the demonstration that the detected alleles segregated with disease in the family, that they were absent in a large group of controls, and/or that they caused reduced cellular abundance of full-length mRNA transcripts, protein products, or mt-ArgRS activity. In spite of that, the functional meaning of p.I9V remains uncertain and, though indirect evidence suggests p.I9V being a hypomorphic allele, we can hypothesize that the modest biochemical defect seen in patient A-01 can be ascribed largely to the c.1586+3A>T change. Modeling of mutations in the yeast orthologue MSR1 showed that MSR1-R531H (= RARS2-R469H) strain could not sustain respiratory growth even when expressed from the relatively strong CYC1 promoter. Conversely, the MSR1-W302R mutant (=RARS2-W241R), retained significant residual activity suggesting its limited contribution to the severe biochemical impairment seen in C-01. Nonetheless, it must be said that yeast modeling of human mutations might not be sufficiently sensitive to disclose the effects of relatively mild, hypomorphic alleles. A similar finding has been noted while modeling in yeast the “common” p.E140K mutation in SCO2 associated with low complex IV in muscle (Dickinson et al 2000), or when testing mutations in genes encoding members of the SBDS protein family (Shammas et al 2005). It is possible that a certain degree of residual activity of mt-ArgRS is essential for embryonic brain development but these aspects need to be further investigated. Published data in zebrafish (Kasher et al 2011) and our own mRNA expression studies (Supplementary Fig. 3) appear to be consistent with this hypothesis.
Lending further evidence to the heterogeneity of clinical presentation of infants with mitochondrial encephalopathies, our work has expanded the array of disease-causing variants in RARS2, offering clues to recognize further similar patients. Nonetheless, a number of questions remained unanswered. Among those, it seems clear that clinical severity cannot be ascribed entirely to the identified mutations nor to their effects on mt-ArgRS or RC activities in muscle (Rankin et al 2010; Glamuzina et al 2011). Unpredictable effects of mitochondrial ARSs mutations on RC activities have already been noticed (Scheper et al 2007; Chrzanowska-Lightowlers et al 2011) and might depend on different tissue expression of tRNAs or ARSs between muscle and brain. Alternatively, mt-ArgRS (or other ARSs) dysfunction may have deleterious consequences in certain neuronal cells only, or mutations might affect functions (i.e., cell signaling, promoting rRNA biogenesis, regulate transcription) other than tRNA charging and protein synthesis (Hausmann and Ibba 2008), at least in neurons (Antonellis and Green 2008; Brown et al 2010). All these concerns need to be addressed in order to better explain genotype-phenotype correlations.
References
Agamy O, Ben-Zeev B, Lev D et al (2010) Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet 87:538–544
Antonellis A, Green ED (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 9:87–107
Ben-Zeev B, Hoffman C, Lev D et al (2003) Progressive cerebellocerebral atrophy: a new syndrome with microcephaly, mental retardation, and spastic quadriplegia. J Med Genet 40:e96
Brown MV, Reader JS, Tzima E (2010) Mammalian aminoacyl-tRNA synthetases: cell signaling functions of the protein translation machinery. Vasc Pharmacol 52:21–26
Budde BS, Namavar Y, Barth PG et al (2008) tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 40:1113–1118
Cassandrini D, Calevo MG, Tessa A et al (2006) A new method for analysis of mitochondrial DNA point mutations and assess levels of heteroplasmy. Biochem Biophys Res Commun 342:387–393
Cassandrini D, Biancheri R, Tessa A et al (2010) Pontocerebellar hypoplasia: clinical, pathologic, and genetic studies. Neurology 75:1459–1464
Chang GG, Pan F, Yeh C, Huang TM (1983) Colorimetric assay for aminoacyl-tRNA synthetases. Anal Biochem 130:171–176
Chang GG, Pan F, Lin YH, Wang HY (1984) Continuous spectrophotometric assay for aminoacyl-tRNA synthetases. Anal Biochem 142:369–372
Chrzanowska-Lightowlers ZM, Horvath R, Lightowlers RN (2011) 175th ENMC International Workshop: mitochondrial protein synthesis in health and disease, 25-27th June 2010, Naarden, The Netherlands. Neuromuscul Disord 21(2):142–147
Dickinson EK, Adams DL, Schon EA, Glerum DM (2000) A human SCO2 mutation helps define the role of Sco1p in the cytochrome oxidase assembly pathway. J Biol Chem 275:26780–26785
DiMauro S (2011) A history of mitochondrial diseases. J Inherit Metab Dis 34:261–276
Edvardson S, Shaag A, Kolesnikova O et al (2007) Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 81:857–862
Enriquez JA, Attardi G (1996) Analysis of aminoacylation of human mitochondrial tRNAs. Methods Enzymol 264:183–196
Friedman SD, Shaw DW, Ishak G, Gropman AL, Saneto RP (2010) The use of neuroimaging in the diagnosis of mitochondrial disease. Dev Disabil Res Rev 16:129–135
Glamuzina E, Brown R, Hogarth K, Saunders D, Russell-Eggitt I, Pitt M, de Sousa C, Rahman S, Brown G, Grunewald S (2011) Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J Inherit Metab Dis. Nov 16
Hausmann CD, Ibba M (2008) Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiol Rev 32:705–721
Hom XB, Lavine JE (2004) Gastrointestinal complications of mitochondrial disease. Mitochondrion 4:601–607
Kasher PR, Namavar Y, van Tijn P et al (2011) Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum Mol Genet 20:1574–1584
Ling J, Reynolds N, Ibba M (2009) Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol 63:61–78
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408
Lloyd AJ, Thomann HU, Ibba M, Söll D (1995) A broadly applicable continuous spectrophotometric assay for measuring aminoacyl-tRNA synthetase activity. Nucleic Acids Res 23:2886–2892
McFarland R, Turnbull DM (2009) Batteries not included: diagnosis and management of mitochondrial disease. J Intern Med 265:210–228
McFarland R, Taylor RW, Turnbull DM (2010) A neurological perspective on mitochondrial disease. Lancet Neurol 9:829–840
Mitochondrial Medicine Society's Committee on Diagnosis, Haas RH, Parikh S, Falk MJ et al (2008) The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab 94:16–37
Naito HK (1975) Modification of the Fiske and SubbaRow method for total phospholipid in serum. Clin Chem 21:1454–1456
Namavar Y, Barth PG, Kasher PR et al (2011) Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134:143–156
Nogueira C, Carrozzo R, Vilarinho L, Santorelli FM (2011) Infantile-onset disorders of mitochondrial replication and protein synthesis. J Child Neurol 26(7):866–875
Peleg D, Kennedy CM, Hunter SK (1998) Intrauterine growth restriction:identification and management. Am Fam Physician 58(2):453–460, 466–7
Rankin J, Brown R, Dobyns WB et al (2010) Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am J Med Genet A 152A:2079–2084
Renbaum P, Kellerman E, Jaron R et al (2009) Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 85:281–289
Scheper GC, van der Klok T, van Andel RJ et al (2007) Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 39:534–539
Shammas C, Menne TF, Hilcenko C et al (2005) Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J Biol Chem 280:19221–19229
Tuppen HA, Fattori F, Carrozzo R et al (2008) Further pitfalls in the diagnosis of mtDNA mutations: homoplasmic mt-tRNA mutations. J Med Genet 45:55–61
Wong LJ (2010) Molecular genetics of mitochondrial disorders. Dev Disabil Res Rev 16:154–162
Acknowledgements
This research was supported in part by grants from the Italian Ministry of Heath, the Telethon Foundation Onlus (Mitocon Project GUP09004 to EB and GGP09207 to LS) Fondazione CARIPARO, Padova (to LS), the European Commission projects FP7 LeukoTreat (to EB), and EUROBFNS (to MRC) and grant from the University of Padova to GS.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Shamima Rahman
Maria Roberta Cilio and Marzia Bianchi contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table
(DOC 223 kb)
Supplementary Table
(DOC 40 kb)
Supplementary Fig. 1
(JPEG 45 kb)
Supplementary Fig. 2A
(JPEG 73 kb)
Supplementary Fig. 2B
(JPEG 101 kb)
Supplementary Fig. 3
(JPEG 69 kb)
Rights and permissions
About this article
Cite this article
Cassandrini, D., Cilio, M.R., Bianchi, M. et al. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J Inherit Metab Dis 36, 43–53 (2013). https://doi.org/10.1007/s10545-012-9487-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-012-9487-9