
Abstract
Pontocerebellar hypoplasia type 6 (PCH6) (MIM #611523) is a recently described disorder caused by mutations in RARS2 (MIM *611524), the gene encoding mitochondrial arginyl-transfer RNA (tRNA) synthetase, a protein essential for translation of all mitochondrially synthesised proteins. This case confirms that progressive cerebellar and cerebral atrophy with microcephaly and complex epilepsy are characteristic features of PCH6. Additional features of PCH subtypes 2 and 4, including severe dystonia, optic atrophy and thinning of the corpus callosum, are demonstrated. Congenital lactic acidosis can be present, but respiratory chain dysfunction may be mild or absent, suggesting that disordered mitochondrial messenger RNA (mRNA) translation may not be the only mechanism of impairment or that a secondary mechanism exists to allow some translation. We report two novel mutations and expand the phenotypic spectrum of this likely underdiagnosed PCH variant, where recognition of the characteristic neuroradiological phenotype could potentially expedite genetic diagnosis and limit invasive investigations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pontocerebellar hypoplasia (PCH) is a relatively frequent radiological finding in paediatric neurometabolic practice and denotes a group of disorders (Table 1), for which there are now six phenotypic subtypes (PCH1–6) and five causative genes identified (Renbaum et al. 2009; Budde et al. 2008; Rajab et al. 2003; Zelnik et al. 1996; Durmaz et al. 2009; Namavar et al. 2011a and b; Edvardson et al. 2007). In 2007, PCH type 6 (PCH6) due to mutations in the nuclear gene RARS2 was first described in three Sephardic Jewish siblings (Edvardson et al. 2007), and since that time, there have been two further reports (Rankin et al. 2010; Namavar et al 2011b). RARS2 is a nuclear gene that encodes mitochondrial arginyl-transfer RNA (tRNA) synthetase (mtRARS), one of a family of 36 aminoacyl-tRNA synthetases (ARSs) (Antonellis and Green 2008).
Messenger RNA (mRNA) translation describes the critical step in protein synthesis whereby genetic information in the form of mRNA is recognised by aminoacylated tRNA and converted to a polypeptide chain. The canonical role of the ARS enzymes is to charge a specific tRNA with its cognate amino acid. This occurs via a two-step enzymatic process known as tRNA aminoacylation that uses one adenosine triphosphate (ATP) molecule to activate the amino acid and transfer it to its tRNA, ready for use in mRNA translation. In addition to the aminoacylation catalytic domain, which is highly conserved from bacteria to humans, most ARSs have a less conserved anti-codon-binding domain and some have additional editing functions. The ARS genes are ubiquitously expressed in all tissues. The ARSs for glycine (GARS), lysine (KARS) and glutamine (QARS) are active in both cytoplasm and mitochondria; however, 17 ARSs are active solely in the mitochondria and 16 solely in the cytoplasm (Antonellis and Green 2008; Guo et al. 2010; Hausmann and Ibba 2008). The 20 ARSs active in the mitochondria are vital for the synthesis of all 13 mitochondrial DNA (mtDNA) encoded proteins. Thus, all complexes of the respiratory chain—other than complex II, which is purely nuclear in origin (Rahman and Hanna 2009)—should be affected by disruption to mtRNA aminoacylation. Accordingly, a multisystem severe phenotype is predicted. The defining characteristics of PCH6 initially described, however, included PCH, epilepsy, cerebral atrophy and multiple respiratory chain enzyme (RCE) defects without evidence of multiorgan disease (Edvardson et al. 2007). In a second report, it was demonstrated that RCE activity may be normal (Rankin et al. 2010), and more recently, a patient was described who at autopsy was found to have had anterior horn-cell disease in addition to PCH, a finding typically associated with PCH1 (Table 1) (Namavar et al. 2011b). Our report of PCH6 due to novel mutations in RARS2 confirms the primary neurodegenerative nature of this disease, expands the clinical and molecular spectrum of this newly described PCH variant and demonstrates the overlapping features within each PCH subtype.
Patient summary
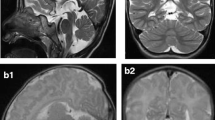
The proband is the first child to nonconsanguineous British Caucasian parents who was delivered at 40 weeks’ gestation after an uncomplicated pregnancy. Birth weight was 2.92 kg (9th percentile) and head circumference 32 cm (0.4th percentile). Physical examination was normal. At 16 h of age, she developed hypotonia and encephalopathy associated with hypoglycaemia, raised anion gap and metabolic acidosis with unrecordably high lactate. In intensive care, she remained unresponsive; acidotic and arterial lactate was 14 mmol/L. Creatine kinase was mildly elevated, and there was mild tubulopathy. Liver function tests were deranged, with abnormal coagulation profile. Workup for a treatable inborn error of metabolism and bacterial or viral pathogens was negative. Initial supportive evidence of mitochondrial dysfunction included high concentrations of proline and alanine in plasma, increased excretion of lactate and pyruvate in urine and reduction in homovanillic acid to 5-hydroxyindoleacetic acid ratio in cerebrospinal fluid (CSF) (Table 2). A muscle biopsy taken at day nine of life showed normal histology. Spectrophotometric assays of RCE activity (as described by Hargreaves et al. 2002) revealed mild reduction of cytochrome oxidase (complex IV) activity relative to citrate synthase (0.011, normal 0.014–0.034). Activities of complex I [nicotinamide adenine dinucleotide (NADH) ubiquinone reductase] and complexes II + III (succinate cytochrome c reductase) were normal (Table 2). Initial magnetic resonance imaging (MRI) of the brain revealed a small pons and dysplastic cerebellum with large bilateral cysts. A small lactate peak was resolved on MR spectroscopy (Fig. 1a, d and g).

A-I: a–c Axial T2-weighted magnetic resonance (MR) images of the brain taken at 5 days, 6 weeks and 7 months of age showing progressive loss of cerebral volume with delayed myelination and relative sparing of the basal ganglia. d–f Coronal T1-weighted MR images of the brain at 5 days, 6 weeks and 7 months of age demonstrating a butterfly pattern of cerebellar hypoplasia with bilateral cysts (+). There is progressive cerebellar volume loss that is most marked between 5 days and 6 weeks but is out of proportion to the degree of supratentorial brain atrophy. g–i Sagittal T1-weighted MR images of the brain at 5 days, 6 weeks and 7 months demonstrating marked progressive cerebral atrophy with progressive thinning of the pons (*), corpus callosum (**) and some degree of optic nerve atrophy (***), particularly between the first two scans
By day 3, the patient was extubated and blood biochemistry had normalised. At 10 days of age, she had a structurally normal eye examination, she was mildly hypotonic but alert, she fed orally and was discharged from hospital. At 4 weeks of age, she re-presented with status epilepticus requiring reintubation. She had frequent jerks, spasticity and fluctuating consciousness level. Her head circumference was 35 cm, now well below the 0.4th percentile for age. There were no biochemical abnormalities detected in blood and urine. Electroencephalogram (EEG) showed severely abnormal background with long periods of marked attenuation, interrupted by sharpened slow activities. Repeat MR imaging of the brain showed remarkable loss of cerebral and cerebellar volume with thinning of the corpus callosum and optic nerves. The basal ganglia remained relatively spared (Fig. 1b, e and h). Over the following 6 months she made no developmental progress, had poor visual attention, no social smiling and continued to have a severe combined seizure and movement disorder. After recovery from prolonged status epilepticus, repeat EEG showed bilateral frontocentral epileptiform discharges, with left-sided predominance. The background showed bilateral anterior more than posterior slowing, suggesting underlying cerebral dysfunction. Repeat brain MR imaging showed minimal worsening (Fig. 1c, f and i). At 12 months, she had near-continuous jerks interrupted by clinically correlated, electrographic seizures, disturbing sleep and required frequent sedation. She was almost entirely dependent on nasogastric feeds, with some pureed food. Auditory and flash visual evoked potentials were absent, with normal flash electroretinograms suggesting profound sensorineural hearing and visual loss. Ophthalmological examination revealed a structurally normal anterior segment, with pupils reacting sluggishly to light, gross pallor of mildly hypoplastic, cupped optic discs with normal intraocular pressure and gross thinning of the nerve fibre layer. This is consistent with secondary optic atrophy associated with loss of function of her anterior vision pathways.
At 24 months of age, she has severe microcephaly and had no motor or cognitive development. She rarely cries and has no purposeful movements. Her jerks are less continuous and are elicited by external stimuli. She has frequent tonic–clonic seizures that are resistant to multiple medications. She is entirely dependent on gastrostomy feeding for nutrition. Her blood and urine lactate have remained normal.
Molecular diagnosis
The appearance of PCH, congenital lactic acidosis and the presence of mild complex IV deficiency led to the suspicion of PCH6 secondary to RARS2 mutations. Using standard methods, complementary DNA (cDNA) and genomic DNA were prepared from cultured fibroblasts from the patient and both parents, amplified by polymerase chain reaction (PCR) and sequenced. Primer pairs for the various PCR are available on request. Initial analysis of cDNA revealed a heterozygous missense mutation, c.1211T>A, M404K, and a heterozygous three base pair (bp) deletion after position c.471 resulting in deletion of lysine 158. Methionine 404 is highly conserved in the RARS2 gene, down through vertebrates, invertebrates to some fungi. The substitution by lysine has not been observed in sequence analysis of normal individuals or other patients suspected of having mutations in the RARS2 gene, nor is it represented in the Single Nucleotide Polymorphism data base (dbSNP). The potential significance of the substitution was analysed using the Sorting Intolerant from Tolerant (SIFT) and PolyPhen programs (Ng and Henikoff 2003; Ramensky et al. 2002) and both predicted that the change would not be tolerated. The human and yeast cytoplasmic and mitochondrial RARS enzymes are members of the class I aminoacyl-tRNA synthetases and share significant sequence homology (supplementary Fig. 1). As the structure of the yeast cytoplasmic enzyme is available (Cavarelli et al. 1998; Delagoutte et al. 2000), it is possible to assess the likely consequences of the mutation based on this structure. Methionine 404 is predicted to be located in helix 15, part of additional domain 2 (Cavarelli et al. 1998). This helix is specifically involved in binding the anticodon loop of tRNA and undergoes significant conformational change on tRNA binding (Delagoutte et al. 2000). This change results in modification of the two signature motifs, which are characteristic of the active site of class I enzymes, and are of key functional importance. Insertion of a positively charged amino acid residue into this helix is very likely to be deleterious.
It is not possible to analyse the consequences of the deletion of lysine 158 using available software packages. However, this residue is also highly conserved and is within a short sequence of amino acids, which is also conserved in both the mitochondrial and cytoplasmic RARS enzymes (supplementary Fig. 1). Based on the structure of the yeast cytoplasmic enzyme, it would be located in helix 6, which makes up part of the active site, and would be just downstream of the conserved HIGH signature sequence of class I synthetases. The loss of a positively charged residue is predicted to disrupt the helix and alter interactions with adjacent parts of the protein in this critical region and is therefore expected to impact activity. This deletion has not been identified in the RARS2 gene of any other normal individuals or patients, nor is it recorded in databases of human sequence variation.
Both mutations were initially identified in cDNA. The heterozygous maternal mutant allele (M404K) was confirmed directly by genomic DNA sequencing; however, the 3-bp deletion was initially not detected in genomic DNA from either the patient or her father. This was subsequently found to be due to a novel intronic base substitution linked to the mutant allele in one of the primer binding sites, a C > A substitution 167 bp upstream of the 5’ end of exon 7. The heterozygous deletion was revealed following primer redesign. This intronic substitution has not been identified in any other individuals but is not considered of functional significance.
Discussion
We present the fourth report of PCH6 secondary to mutations in RARS2. As was the patient described by Rankin et al. (2010), our patient is a British Caucasian child born to nonconsanguineous parents. Both patients were compound heterozygous for two novel mutations, thus expanding the mutation spectrum of this seemingly rare disorder (Table 3).
Reduced mitochondrial RCE activity without significant biochemical or neuroradiological features typical of a mitochondrial disorder was initially considered a hallmark feature of PCH6 (Edvardson et al. 2007). Our patient had only mildly reduced muscle complex IV activity, and the fourth case reported had no abnormality detected. Nonetheless, both British patients had severe but transient congenital lactic acidosis, a feature not seen in the first reported cases (Table 3). Our case is the first reported to have transient liver dysfunction, mild renal tubulopathy and low levels of biogenic monoamine metabolites HVA and 5-HIAA, which could also be explained by mitochondrial dysfunction (Garcia-Cazorla et al. 2008).
A common clinical feature of the other PCH subtypes is complex, treatment-resistant epilepsy. In the three patients described by Edvardson et al (2007), patient II-2 had intractable seizures with generalised discharges, some side preference and attenuation of the activities on EEG, similar in description to the changes observed in our patient. Their patient II-4 did not appear to have seizures, but patient II-5 developed these at 4 months of age. An EEG at 3 weeks of age was normal, but there is no report of any further recordings. The patient described by Rankin et al. (2010) had myoclonic seizures with epileptiform phenomena on EEG confirmed at 1 week of age (Table 3). From these descriptions, it is not possible to delineate a shared distinguishing electrophysiological feature.
Our case demonstrates the consistent neuroimaging finding of marked flattening or butterfly shape (Namavar et al. 2011b) of the cerebellum and cerebral volume loss, which appears to be progressive—in our patient, rapidly progressive—and is associated with concordant microcephaly. Cerebral and cerebellar atrophy is also observed in PCH3. In addition, these patients have corpus callosum thinning and early-onset optic atrophy (Rajab et al. 2003; Zelnik et al. 1996; Durmaz et al. 2009), which was observed for the first time in PCH6 in our patient (Table 3). Cerebellar cysts seen are a rare neuroradiological finding recently reported in a child with PCH2 due to TSEN54 mutations (Namavar et al. 2011b). The combination has also been described in association with bilateral frontoparietal polymicrogyria due to mutations in GPR56 (Chang et al. 2003; Piao et al. 2005). Cysts are also associated with congenital muscular dystrophies due to defects in dystroglycan glycosylation (Barkovich 1998; Clement et al. 2008). The specific neuroradiological changes seen in PCH6 are of interest, as they allow the potential for rapid recognition and genetic diagnosis, thus bypassing the need for muscle biopsy.
There are nine reported diseases caused by mutations in ARS genes. Autosomal dominant mutations in two cytoplasmic ARS genes—YARS (Jordanova et al. 2006) and AARS (Latour et al. 2010)—and one bifunctional ARS gene, GARS (Antonellis et al. 2003), result in a Charcot-Marie-Tooth (CMT) phenotype (Table 1). The six mitochondrial ARS diseases described to date are recessively inherited. Compound heterozygous mutations in DARS2 encoding aspartyl-tRNA synthetase result in leukoencephalopathy and brainstem and spinal cord lesions with elevated lactate (LBSL) in the CNS but without systemic manifestations of mitochondrial dysfunction (Scheper et al. 2007). In contrast, homozygosity for a single mutation c.156C>G (p.F52L) in YARS2 has been found in two unrelated families with myopathy, lactic acidosis and sideroblastic anaemia (MLASA) (Riley et al. 2010), and homozygous mutations in SARS2 are associated with renal tubulopathy, hyperuricemia, metabolic alkalosis, pulmonary hypertension and progressive renal failure in infancy (HUPRA syndrome) (Belostotsky et al. 2011). Both of these reported cases are associated with impaired mitochondrial RCE activity in muscle. Compound heterozygous mutations in HARS2 have been found in the original kindred described in 1979 with Perrault syndrome or ovarian dysgenesis and progressive sensorineural hearing loss (Pierce et al. 2011). Most recently, mutations in AARS2 have been identified in three patients with severe infantile- and prenatal-onset cardiomyopathy and combined RCE deficiency in heart muscle (Götz et al. 2011).
The pivotal role of ARSs in mRNA translation and their ubiquitous expression would suggest that mutations in these genes should have widespread phenotypic consequences. This is not the case; and the reason, in particular for the predominantly neurological phenotype in RARS2, DARS2 and the cytoplasmic ARS diseases, is not well understood. When a number of different human and mouse mutations in the glycl-tRNA synthetase gene were introduced into the yeast orthologue, complementation studies revealed no impairment in protein synthesis (Stum et al. 2011). Some cytoplasmic ARS have incorporated secondary domains with roles that are unrelated to aminoacylation (Hausmann and Ibba 2008; Guo et al. 2010). It is conceivable that mutations affect these noncanonical functions (Stum et al. 2011). However, these have not been identified in mitochondrial ARS and the majority of mutations associated with CMT phenotype occur in the catalytic or anticodon domains essential for aminoacylation. Neuronal cells may potentially have an increased requirement for protein synthesis (Antonellis and Green 2008; Stum et al. 2011). A reduction, but not complete ablation, of the efficiency of tyrosyl-tRNA synthetase aminoacylation is associated with mutations in YARS2 (Riley et al. 2010). A defect in aminoacylation was also demonstrated in one of the patients with mutations in RARS2 (Edvardson et al. 2007) and in the patients with SARS2 mutations (Belostotsky et al. 2011). Although functional studies on the mutant enzyme have not been performed in the case presented here, the predicted alterations to the protein structure are expected to affect catalytic function directly. Based on homology with the yeast cytoplasmic arginyl-tRNA synthetase, the deleted lysine residue 158 would be located within the active site domain of the enzyme itself, whereas the lysine residue substituting for methionine 404 would be in a helical segment in the C-terminal additional domain 2 (Cavarelli et al. 1998). This helical segment undergoes a large conformational change when the enzyme binds the anticodon loop of the tRNA, and this structural change is a key feature of the catalytic mechanism. Hence, the clinical and biochemical manifestations of PCH6 and the other mitochondrial ARS disorders appear to be due to reduction in aminoacylation and, by extension, a reduction in mRNA translation and protein synthesis.
Significant clinical overlap exists between the six PCH phenotypes, implying a similar underlying pathogenesis. TSEN2, TSEN34, and TSEN54 encode subunits of the cytoplasmic tRNA splicing endonuclease. Mutations in these genes are associated with PCH2, PCH4 (Budde et al. 2008; Namavar et al. 2011b) and PCH5 phenotypes (Namavar et al. 2011a). The tRNA splicing endonuclease complex is essential for mRNA translation within the cytoplasm, as it removes the intron from tRNA to form mature tRNA. It is required for the synthesis of all cytoplasmic proteins; thus, disturbed endonuclease function may indirectly interfere with mitochondrial protein synthesis via the abnormal translation of proteins such as the mitochondrial ARSs or via disruption of transport of such proteins into the mitochondria. However, there are no reports of widespread mitochondrial dysfunction in patients with TSEN2, TSEN34 and TSEN54 mutations. PCH1 is thought to be associated with mutations in VRK1, a serine-threonine kinase that phosphorylates a number of transcription factors and is an essential regulator of cell division (Valbuena et al. 2011). Interestingly, the putative locus 7q11-21 mapped in a family with PCH3 phenotype (Rajab et al. 2003) contains one gene, EIF4H, which is a eukaryote translation initiation factor that combines with two other initiation factors to form a helicase that unwinds mRNA secondary structures in preparation for translation (Parsyan et al. 2011). This locus also contains two zinc-finger genes that are important in transcription initiation, but no diseases have been ascribed to them (Table 1).
An alternative mechanism proposed for ARS-related disorders is the accumulation of a neurotoxic substance, such as mutant ARS aggregates or proteins with aberrant amino acid residues (Stum et al. 2011; Antonellis and Green 2008). Accordingly, altered function of arginyl-tRNA synthetase and the tRNA splicing endonuclease complex may lead to accumulation of unprocessed tRNA that may, in turn, result in abnormal cerebellar development, neurotoxicity and subsequent neurodegeneration. Whatever the mechanism, the paucity of multiorgan disease associated with PCH due to TSENopathies (Namavar et al 2011a) or RARS2 mutations suggests there must be a secondary mechanism that allows for compensation of this defect, at least in peripheral tissues.
In summary, this case confirms that mutations in RARS2 result in progressive and potentially rapid cerebral and cerebellar atrophy, microcephaly and complex epilepsy. Cerebellar cysts, optic atrophy and severe movement disorder seen in other PCH subtypes have not been previously reported. The occurrence in two nonconsanguineous children with compound heterozygosity suggests RARS2 mutations may be more prevalent than is recognised. As there is overlap between PCH subtypes, RARS2 sequencing with complementary and genomic DNA in all patients with progressive cerebral and cerebellar atrophy would seem prudent, particularly but not exclusively if there is clinical or biochemical evidence of mitochondrial RCE dysfunction. Recognition of this phenotype is important, as this represents one of the few congenital lactic acidoses in which a specific genetic diagnosis may be suggested by MRI changes.
References
Antonellis A, Green ED (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 9:87–107
Antonellis A, Ellsworth RE, Sambuughin N et al (2003) Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72:1293–1299
Barkovich JA (1998) Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol 19:1389–1396
Belostotsky R, Ben-Shalom E, Rinat C et al (2011) Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet 88:193–200
Budde BS, Namavar Y, Barth PG et al (2008) tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 40:1113–1118
Cavarelli J, Delagoutte B, Eriani G et al (1998) L-arginine recognition by yeast arginyl-tRNA synthetase. EMBO J 17:5438–5448
Chang BS, Piao X, Bodell A et al (2003) Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann Neurol 53:596–606
Clement E, Mercuri E, Godfrey C et al (2008) Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 64:573–582
Delagoutte B, Moras D, Caravelli J (2000) tRNA aminoacylation by arginyl-tRNA synthetase: induced conformations during substrates binding. EMBO J 21:5599–5610
Durmaz B, Wollnik B, Cogulu O et al (2009) Pontocerebellar hypoplasia type III (CLAM): extended phenotype and novel molecular findings. J Neurol 256:416–419
Edvardson S, Shaag A, Kolesnikova O et al (2007) Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 81:857–862
Garcia-Cazorla A, Duarte S, Serrano M et al (2008) Mitochondrial diseases mimicking neurotransmitter defects. Mitochondrion 8:273–278
Götz A, Tyynismaa H, Euro L et al (2011) Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 88:635–642
Guo M, Yang XL, Schimmel P (2010) New functions of aminoacyl tRNA synthetases. Nat Rev Mol Cell Biol 11:668–674
Hargreaves P, Rahman S, Guthrie P et al (2002) Diagnostic value of succinate ubiquinone reductase activity in the identification of patients with mitochondrial DNA depletion. J Inherit Metab Dis 25:7–16
Hausmann CD, Ibba M (2008) Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiol Rev 32:705–721
Jordanova A, Irobi J, Thomas FP et al (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Toot neuropathy. Nat Genet 38:197–202
Latour P, Thauvin-Robinet C, Baudelet-Méry C et al (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 86:77–82
Namavar Y, Chitayat D, Barth PG et al (2011a) TSEN54 mutations cause pontocerebellar hypoplasia type 5. Eur J Hum Genet 19:724–726
Namavar Y, Barth P, Kasher P et al (2011b) Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134:143–156
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814
Parsyan A, Svitkin Y, Shahbazian D et al (2011) mRNA helicases: the tacticians of translational control. Nat Rev Mol Cell Biol 12:235–245
Patel MS, Becker LE, Toi A et al (2006) Severe, fetal-onset form of olivopontocerebellar hypoplasia in three sibs: PCH type 5? Am J Med Genet 140A:594–603
Piao X, Chang BS, Bodell A et al (2005) Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol 8:680–687
Pierce SB, Chisholm KM, Lynch ED et al (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 108:6543–6548
Rahman S, Hanna MG (2009) Diagnosis and therapy in neuromuscular disorders: diagnosis and new treatments in mitochondrial diseases. J Neurol Neurosurg Psychiatry 80:943–953
Rajab A, Mochida GH, Hill A et al (2003) A novel form of pontocerebellar hypoplasia maps to chromosome 7q11-21. Neurology 60:1664–1667
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30:3894–3900
Rankin J, Brown R, Dobyns WB et al (2010) Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am J Med Genet A 152A:2079–2084
Renbaum P, Kellerman E, Jaron R et al (2009) Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 85:281–289
Riley LG, Cooper S, Hickey P et al (2010) Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet 87:52–59
Scheper GC, van der Klok T, van Andel RJ et al (2007) Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 39:534–539
Stum M, McLaughlin HM, Kleinbrink EL et al (2011) An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Mol Cell Neurosci 46:432–443
Valbuena A, Sanz-García M, López-Sánchez I et al (2011) Roles of VRK1 as a new player in the control of biological processes required for cell division. Cell Signal. Apr 14 [Epub ahead of print]
Zelnik N, Dobyns WB, Forem S et al (1996) Congenital pontocerebellar atrophy in three patients: clinical, radiologic and etiologic considerations. Neuroradiology 38:684–687
Acknowledgement
We acknowledge our patient’s family for allowing us to publish her medical information and MRI images. Shamima Rahman is funded by Great Ormond Street Hospital Children’s Charity.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: John Christodoulou
Competing interest: None declared.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
(DOC 95 kb)
Rights and permissions
About this article
Cite this article
Glamuzina, E., Brown, R., Hogarth, K. et al. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2 . J Inherit Metab Dis 35, 459–467 (2012). https://doi.org/10.1007/s10545-011-9413-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9413-6