
Abstract
Treatment with tetrahydrobiopterin (BH4), the natural cofactor of phenylalanine hydroxylase (PAH), can reduce blood phenylalanine (Phe) levels in patients with BH4-responsive phenylketonuria (PKU). A number of studies has reported on the short-term BH4 treatment of patients with PKU, but long-term data are lacking. Here, we describe the effects of long-term treatment with BH4 on 16 patients, who showed a >28% reduction in blood Phe following testing for BH4 overload. The mean dose of BH4 was 16 mg/kg body weight (range 5–36 mg/kg body weight). The mean treatment duration was 56 months (range 24–110 months). Of 16 patients, 14 achieved long-term Phe control with BH4 treatment, with a mean blood Phe concentration of 321 ± 236 µmol/l. The mean decrease from baseline in blood Phe levels in these 14 patients was 54.6%. Of the seven patients who required continued dietary restriction, Phe intake increased from 200–300 mg/day to 800–1000 mg/day. Factors that may cause fluctuation of Phe levels in BH4-treated patients include patients’ PAH genotype, Phe intake, changes in protein catabolism or anabolism, and periods of illness or infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Classical phenylketonuria (PKU; OMIM 262600) is a rare metabolic disorder that usually results from a deficiency of a liver enzyme known as phenylalanine hydroxylase (PAH; EC 1.14.16.1). This enzyme deficiency leads to elevated levels of the amino acid phenylalanine (Phe) in the blood and other tissues. The untreated state is characterized by mental retardation, microcephaly, delayed speech, seizures, eczema and behaviour abnormalities.
PKU can be divided into three groups: mild hyperphenylalaninaemia (MHPA), in which blood Phe concentrations are less than 600 µmol/l; mild PKU, in which blood Phe levels are between 600 µmol/l and 1,200 µmol/l, and classical PKU, in which blood Phe levels exceed 1,200 µmol/l if left untreated. Adherence to a low-Phe diet from birth is effective in preventing mental retardation, but it is very restrictive and limits patient and family quality of life. In addition, such a restrictive diet is difficult to control in children, and adherence can be poor. There is a consensus in Germany that treatment should be given only to patients with mild and classical PKU (blood Phe >600 µmol/l) (Burgard et al. 1999).
In a subgroup of responsive patients, high doses of tetrahydrobiopterin (BH4), a natural cofactor for PAH, stimulated residual enzyme activity and reduced blood Phe levels (Bélanger-Quintana et al. 2005; Kure et al. 1999; Lambruschini et al. 2005). The molecular mechanisms responsible for BH4-responsiveness in patients with PKU are multifactorial and depend upon the exact mutations present in the PAH gene (Zurflüh et al. 2008). Following the completion of phase III studies, a synthetic formulation of BH4, sapropterin dihydrochloride (Kuvan®, BioMarin Pharmaceutical Inc., Novato, USA), was recently approved for use in conjunction with a Phe-restricted diet for the treatment of patients with hyperphenylalaninaemia due to BH4-responsive PKU (Lee et al. 2008; Levy et al. 2007).
Considering the life-long nature of PKU, the vast majority of studies investigating BH4 treatment have been performed over a relatively short time; longer-term studies are required to monitor the occurrence of any adverse events and to optimize treatment strategies further, including the relaxation of a Phe-restricted diet (Trefz et al. 2005). However, a number of inherent limitations involved in the long-term evaluation of BH4 treatment in patients with PKU have made this a difficult task.
-
1.
Heterogeneous patient group
The patient group in PKU is a heterogeneous one. Patients require continuous monitoring throughout their lifetimes, and, thus, any study group may encompass infants, children, adolescents and adults. Additionally, dietary restrictions and dietary compliance vary greatly between individuals, with some patients taking a relaxed approach to their diet and others adhering strictly to recommended guidelines. Adherence to treatments and regular Phe monitoring also varies between individuals.
-
2.
No standardized protocol for BH4 treatment
The lack of a standardized protocol for BH4 treatment and subsequent dietary modifications results in an individual response to therapy. This makes general comparisons of long-term treatment and subsequent recommendations difficult to undertake and apply.
-
3.
Increased protein intake following initiation of BH4 treatment
The switch from purely diet-controlled disease management to drug therapy may lead to increased dietary protein intake, as patients feel that BH4 treatment will automatically allow them to eat a normal diet. Whereas successful treatment with BH4 does allow for increased dietary freedom in many patients, careful monitoring is required to ensure that blood Phe levels remain under control.
-
4.
Phe tolerance decreases with age in classical PKU
A natural decrease in Phe tolerance with increased age means that treatment strategies need adjustment to maintain blood Phe concentrations at an acceptable level.
This study aimed to increase the understanding of the long-term effects of treatment with BH4 in patients with PKU in order to further optimize treatment strategies.
Methods and study design
All patients must have received treatment for PKU in accordance with treatment guidelines: infants and children with Phe levels >600 µmol/l; adolescents and adults with Phe levels >1,200 µmol/l (Burgard et al. 1999). In addition, a clear response to BH4 treatment was required, with a >30% reduction in blood Phe levels evident after either an acute BH4-overload test (20 mg/kg body weight over 24 h) or long BH4-overload test (20 mg/kg body weight over 8 days). Informed consent was provided by all participants.
Patients attended regular assessments (at least twice a year), during which clinical status was assessed and routine blood chemistry analyses were performed. Blood Phe was measured in the first year of life at weekly intervals, twice monthly from the second year, and once a month in adults, using capillary blood drawn onto dried filter paper. Blood Phe levels were measured by tandem mass spectrometry.
Tetrahydrobiopterin was provided from Schircks Laboratories, Switzerland. Patients with identification (ID) numbers 159, 937, 715 (Table 1) were treated by sapropterin provided by BioMarin Pharmaceutical Inc. during the PKU 006 study (Trefz et al. 2009). The tablets were dissolved in a glass of water and taken once in the morning. Patients ID nos. refer to the RAMEDIS database, where all blood phe levels are documented.
Results
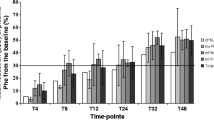
A total of 16 patients were included in the analysis (Table 1). Except for patient, ID no. 159, who had the somewhat lower response of 30%, all patients had a clear response of >30%. The mean treatment dose of BH4 was 16 mg/kg body weight per day. The mean treatment duration was 56 months, and the longest time on treatment was 110 months. Of the 16 patients, 14 (87.5%) achieved long-term Phe control with BH4 treatment (Fig. 1). Two patients (patients 715 and 937), who were non-responders on long-term follow-up, showed high fluctuations in blood Phe levels (Figs. 1, 2). However, these same two patients experienced a sharp decline in blood Phe levels after the start of an 8-day BH4-loading test, a chance measurement at one instant in time corresponding to a pseudo-response on BH4 load. Mean blood Phe levels and fluctuations in blood Phe are shown for BH4 responders in Fig. 3a,b, and typically show levels of blood Phe that lie on or within the upper limit of the Phe treatment recommendation from the National Institutes of Health Consensus Development Panel (NIH) (2001); fluctuations in blood Phe are low. By contrast, blood Phe levels in patients 715 and 937 (Fig. 3c,d, respectively), have initially high fluctuations that gradually decline as blood Phe levels increase, reaching a ‘saturation’ point indicating poor compliance of dietary treatment and no response to BH4 supplementation, especially in patient 715.

Mean blood phenylalanine (Phe) concentrations in 16 patients with phenylketonuria treated with tetrahydrobiopterin (BH4). Circles represent the two non-responders on long-term follow-up: two patients with uncontrolled Phe levels (patients 715 and 937) who were classed as ‘pseudo-responders’. The long solid horizontal line shows the upper limit of Phe treatment, as recommended by the National Institutes of Health Consensus Development Panel (2001). The short-dashed lines show the standard deviation (SD) (±236 µmol/l) of the mean blood Phe level (321 µmol/l, illustrated by the long-dashed line) in BH4-treated patients classed as responders (n = 14)

Long-term blood phenylalanine (Phe) profiles showing high fluctuations in patients a 715 and b 937. For both patients, tetrahydrobiopterin (BH4) loading resulted in a ‘decrease’ in blood Phe (arrows) after an 8-day BH4-loading test; this illustrates a pseudo-response to a BH4 load

Change in blood phenylalanine (Phe) in good tetrahydrobiopterin (BH4) responders: a patient 445 and b patient 230; and BH4 non-responders (pseudo-responders): c patient 715 and d patient 937, during long-term treatment with BH4. Black vertical bars represent mean blood Phe value per year; white vertical bars represent the standard error of estimation. *Consensus level values for blood Phe as recommended by the National Institutes of Health Consensus Development Panel (2001). The thick line under each x-axis indicates the duration of BH4/sapropterin treatment
The mean blood Phe concentration in the 14 responders was 321 ± 236 µmol/l, and the mean decrease in blood Phe levels was 54.6%. Seven patients achieved stable Phe control without dietary Phe restriction, and, of the remaining patients who required continued dietary Phe restriction, six increased their Phe intake from 300 mg/day to 1,000 mg/day; two did not (Table 1).
In one patient with mild PKU (patient 759), there was an increase in blood Phe levels under catabolic conditions and a strictly controlled low-Phe diet (Phe intake 220 mg/day). Blood Phe levels normalized following initiation of BH4 treatment (10 mg/kg per day) in combination with a relaxed diet (Phe intake 500 mg/day). Subsequently, the patient’s body weight and height increased (3rd percentile) to greater than the 50th percentile after relaxation of diet with a higher content of natural protein.
In a second patient who initially responded well to BH4 treatment (patient 494) there was an apparent loss of effectiveness, resulting in increased blood Phe levels. The initial BH4 dose (5 mg/kg per day) was stopped for 5 days, and Phe levels increased; when BH4 was re-introduced, Phe levels decreased. As a result of an infection, Phe levels increased again—Phe control and increased body weight were achieved by an increase in the BH4 dose to 10 mg/kg per day.
In all patients, no side effects related to BH4 treatment were observed. Psychomotor development, as documented by the Hamburg Wechsler Intelligenz-Test für Kinder (HAWIK III), in infants aged 5–6 years was within the normal range.
Discussion
In this long-term follow-up study (which has now been completed), 14 patients responded well to BH4 treatment and blood Phe levels were maintained within guideline values. The two non-responders showed insufficient Phe control according to the diet for life concept (NIH consensus 2001). The mean treatment duration was 56 months, and one patient has now received BH4 therapy for 110 months (over 9 years). In addition, seven out of 14 responsive patients received BH4 monotherapy without dietary Phe restriction, whereas the other seven responsive patients increased their dietary Phe intake. Overall, BH4 treatment was well tolerated, and its effectiveness was maintained with long-term treatment.
Good BH4 responders, as shown in Fig. 3a,b (representing patients 445 and 230, respectively), typically experienced low fluctuations in blood Phe levels that all fell within the target range. However, non-responders (patients 715 and 937; Fig. 3c,d) had long-term, very high blood Phe with low Phe fluctuations because of insufficient Phe control. These two patients, also described as ‘pseudo-responders’, displayed high fluctuations in blood Phe levels between days 1 and 8 of the BH4-loading test. Pseudo-responders were defined as patients who mimic a positive response (>30% decrease in blood Phe) after 8 days following sapropterin loading, in this instance, the report of a chance decline in high fluctuations of blood Phe. However, upon long-term follow-up, both patients showed no response to treatment and typically were characterized by very high blood Phe with low fluctuations.
The pseudo-response observed in these patients might have been caused by a high fluctuation index in blood Phe under unstable treatment conditions. The fluctuation index can be expressed through the standard error of estimation (SEE), which correlates with the severity of PKU expressed as the residual enzyme activity of PAH (r = 0.61, P < 0.001) and intelligence quotient at 9 years of age (r = 0.37, P < 0.05) (Burgard et al. 1996). This suggests that testing for BH4 responsiveness with the 24 h BH4-loading test, or using only two points of measurements between days 1 and 8 on the long BH4-loading test, may not be suitable for dietary restricted patients who have a high fluctuation index for Phe levels (SEE > 250–400 µmol/l) (Fig. 3). It has been shown that a high fluctuation index is associated with mutations or genotypes with low in vitro residual enzyme activity of the PAH system (Burgard et al. 1996). It is possible, therefore, that the underlying PAH genotype of these patients (p.R261Q/p.R243L in Patient 715 and p.R158Q/IVS4 + 5G > T in Patient 937) accounts for their high fluctuation index.
In addition to the two pseudo-responders, individual patient reports in this study highlight the need for the continual monitoring of blood Phe levels as well as the individualization of BH4 treatment throughout the lifetime of the patient.
The varying response observed across the patient group treated with BH4 (e.g. effective BH4 dose, requirements for a Phe-restricted diet) also indicates that a number of factors affect an individual’s response to BH4 treatment. Protein metabolism and infection are two factors thought to affect Phe fluctuation in patients with PKU, together with factors affecting pharmacological parameters (e.g. absorption, distribution, metabolism and drug clearance). As Phe tolerance decreases and blood Phe levels generally increase with age in patients with PKU, age may also be a factor in an individual’s response to treatment.
Conclusion
Long-term treatment with BH4 was effective and well tolerated in the majority of patients with BH4-responsive PKU, without loss of efficacy. Factors that may cause fluctuation in the Phe levels of patients treated with BH4 include PAH genotype, changes in dietary Phe intake, changes in protein catabolism or anabolism, variation in individual pharmacological parameters, patient age, treatment adherence, catabolic conditions such as infections, and dietary compliance. This illustrates the need for BH4 dose adjustment in the long-term and acute illness.
Abbreviations
- BH4 :
-
tetrahydrobiopterin
- EC:
-
Enzyme Commission
- HAWIK:
-
Hamburg Wechsler Intelligenz-Test für Kinder
- MHPA:
-
mild hyperphenylalaninaemia
- OMIM:
-
Online Mendelian Inheritance in Man database
- PAH:
-
phenylalanine hydroxylase
- Phe:
-
phenylalanine
- PKU:
-
phenylketonuria
- RAMEDIS:
-
rare metabolic disease database www.ramedis.de
- SD:
-
standard deviation
- SEE:
-
standard error of estimation
References
Bélanger-Quintana A, García MJ, Castro M et al (2005) Spanish BH4-responsive phenylalanine hydroxylase-deficient patients: evolution of seven patients on long-term treatment with tetrahydrobiopterin. Mol Genet Metab 86(Suppl 1):S61–S66
Burgard P, Rupp A, Konecki DS, Trefz FK, Schmidt H, Lichter-Konecki U (1996) Phenylalanine hydroxylase genotypes, predicted residual enzyme activity and phenotypic parameters of diagnosis and treatment of phenylketonuria. Eur J Pediatr 155(Suppl 1):S11–S15
Burgard P, Bremer HJ, Bührdel P et al (1999) Rationale for the German recommendations for phenylalanine level control in phenylketonuria 1997. Eur J Pediatr 158:46–54
Kure S, Hou DC, Ohura T et al (1999) Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr 135:375–378
Lambruschini N, Pérez-Dueñas B, Vilaseca MA et al (2005) Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab 86(Suppl 1):S54–S60
Lee P, Treacy EP, Crombez E et al (2008) Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet 146A:2851–2859
Levy HL, Milanowski A, Chakrapani A et al (2007) Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 370:504–510
National Institutes of Health Consensus Development Panel (2001) National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics 108:972–982
Trefz FK, Scheible D, Frauendienst-Egger G, Korall H, Blau N (2005) Long-term treatment of patients with mild and classical phenylketonuria by tetrahydrobiopterin. Mol Genet Metab 86(Suppl 1):S75–S80
Trefz FK, Burton BK Longo N, Casanova MM et al (2009) Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 154:700–707
Zurflüh MR, Zschocke J, Lindner M et al (2008) Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Hum Mutat 29:167–175
Acknowledgements
The authors take full responsibility for the content of this article but thank Caudex Medical (supported by Serono Symposia International Foundation) for their assistance in preparing the initial draft of this report and collating the comments of the authors and any other named contributors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Nenad Blau
Reference to electronic databases: EC, OMIM, RAMEDIS
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Trefz, F.K., Scheible, D. & Frauendienst-Egger, G. Long-term follow-up of patients with phenylketonuria receiving tetrahydrobiopterin treatment. J Inherit Metab Dis 33 (Suppl 3), 163–169 (2010). https://doi.org/10.1007/s10545-010-9058-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9058-x