
Summary
Pregnancy management in phenylketonuric women includes continuous dietary control starting before conception, aiming to maintain blood phenylalanine concentrations in a desirable range, irrespective of the fetal genetic PKU status. While the maternal phenylalanine hydroxylase (PAH) genotype will influence metabolic control, an effect of the fetal genetic PKU status on maternal metabolic control during pregnancy has not been described. We monitored three pregnancies of women with classical PKU by dietary protocols of daily phenylalanine intake, phenylalanine blood concentrations, and obstetric care. Patients 1 and 2 carried a heterozygous (not PKU-affected) fetus, while patient 3 was pregnant with a PKU-affected fetus (PAH p.R408W and p.R408W). The expected increase in phenylalanine tolerance during the course of pregnancy was observed in patients 1 and 2 in whom phenylalanine intake could be steadily increased from 400 to 1700 mg/day while phenylalanine blood concentrations remained in the desired range. Gain of body weight was 13.0 and 17.7 kg, respectively. In patient 3, the phenylalanine tolerance did not rise above 600 mg/day, and phenylalanine blood concentrations were above the desired range on several occasions. Caloric intake was therefore encouraged, which led to a weight gain of 20.0 kg. The course of pregnancy was otherwise normal in all three cases, and infants with normal birth weight and head circumference were born. The different phenylalanine tolerance in pregnancies with PKU-affected and non-affected fetuses suggests that PAH genotype and metabolic situation of the fetus influence maternal metabolic control. A phenylalanine tolerance remaining low in the third trimester of pregnancy may indicate fetal PKU.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Present recommendations for pregnancy management in women with PKU include continuous dietary control with blood phenylalanine concentrations between 100 and 250 μmol/L starting before conception (Maillot et al 2007), irrespective of the fetal phenylalanine hydroxylase (PAH) genotype, in order to avoid not only mental retardation but also congenital heart disease (Levy et al 2001). While the maternal PAH genotype will influence metabolic control, the effect of the fetal genetic PKU status on maternal metabolic control during pregnancy has not been described before.
Subjects and methods
Three consecutive pregnancies in three women with classical PKU were managed in our metabolic clinic for adults. Dietary treatment included supplements with a nutritional mixture specifically devised for pregnant PKU patients (P-AM maternal) and p-am Anamix (both from SHS, Heilbronn, Germany). The daily dose of the supplement was adjusted in relation to the patients’ tolerance of natural protein and was taken with meals in 4–5 single doses per day. The patients were monitored by dietary protocols (allowing the calculation of daily phenylalanine intake), by determination of phenylalanine blood concentrations, and by obstetrical care that included fetal ultrasound examinations in the 10th and 21st weeks. Patient 3 had an additional ultrasound examination in the 28th week. The case histories are presented below. For details such as maternal weight gain during pregnancy and the status of the newborns, see Table 1. At our centre, blood phenylalanine concentrations between 60 and 240 μmol/L, starting before conception, were recommended at the time of this study, in close accordance with recommendations of the German Working Group for Paediatric Metabolic Disorders (Bremer et al 1997).
Patient 1 was a 30-year-old PKU woman with the PAH mutations p.I94del (c.284_286delTCA in exon 3) and p.P281L (c.842C>T in exon 7). She was an early-diagnosed, compliant, well functioning PKU patient. At the age of 23 years, she had a prior diet-controlled pregnancy that resulted in a completely normal child. Here, we report on her second pregnancy carrying a heterozygous (not PKU-affected) fetus. The present pregnancy was unplanned; dietary management was started two weeks after her last menstrual period.
Patient 2 was a 27-year-old woman with the PAH mutations p.I65T (c.194T>C in exon 3) and p.R408W (c.1222C>T in exon 12), also carrying a heterozygous (not PKU-affected) fetus. She had been an early-diagnosed PKU patient, but her metabolic control during childhood and adolescence was not satisfactory. At the age of 19 years, she had a spontaneous abortion. At the age of 24 years, she had a pregnancy without any dietary monitoring and delivered a microcephalic, mentally retarded child with hypoplastic corpus callosum. The present pregnancy, with a new partner, was started with high motivation and after a successful period of dietary training with phenylalanine blood levels in the recommended range.
Patient 3 was a 26-year-old woman with the PAH mutations p.R408W (c.1222C>T in exon 12) and p.R261Q (c.782G>A in exon 7). She was an early-diagnosed, well managed PKU patient who had never stopped her PKU diet. She became a nurse and met her present partner at a meeting for PKU patients. At the age of 23 years she had an intentional abortion in the 15th week of pregnancy. The present pregnancy was planned after intensive preparation and a successful period of dietary training. As the child’s father was also a PKU patient (homozygous for PAH p.R408W), she was carrying a PKU-affected fetus (homozygous for PAH p.R408W).
Results
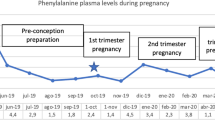
The increase in phenylalanine tolerance (Hyanek et al 1988) during the course of pregnancy differed remarkably between the three women. In patients 1 (Fig. 1, top) and 2 (Fig. 1, centre), phenylalanine intake could be increased from 400 to 1700 mg/day while phenylalanine blood levels remained in the desired range. Gain of body weight was 13 and 17.7 kg, respectively. In patient 3 (Fig. 1, bottom), phenylalanine intake could not be increased above 400–600 mg/day without risking high phenylalanine blood concentrations. Caloric intake (phenylalanine-free) was therefore encouraged, resulting in a weight gain of 20.0 kg. At birth, however, the newborn’s weight, when expressed as a proportion of maternal weight gain during pregnancy, was only 16% and lower than in the pregnancies of patients 1 and 2 (Table 1).

Course of three consecutive PKU pregnancies. Phenylalanine blood levels are shown as black dots; the recommended upper limit of blood phenylalanine during pregnancy is marked by a white line; the daily phenylalanine uptake is depicted as white squares. The day of birth is marked by an arrow. Top: Pregnancy with a fetus heterozygous for PKU (patient 1). Centre: Pregnancy with a fetus heterozygous for PKU (patient 2). Bottom: Pregnancy with a fetus homozygous for PKU (patient 3)
The course of pregnancy was otherwise normal in all three cases. Findings in repeated fetal ultrasound examinations were normal. In the fetus of patient 3, however, the head size, as estimated by measuring the biparietal and fronto-occipital diameters, declined from between the 50th and 95th centiles in the 21st week to between the 5th and 50th centiles in the 28th week. The newborn infants had normal birth weight, length, and head circumference. The development of all children has been normal over a follow-up period of 3 years.
Discussion
While the birth of a phenylketonuric child to a phenylketonuric mother has sporadically been mentioned in literature, dating back to the very first descriptions of the maternal PKU syndrome (Levy 2003), the significance of fetal phenylalanine metabolism on maternal phenylalanine tolerance has remained unclear. In two pregnant PKU patients carrying heterozygous (not PKU-affected) fetuses, dietary management was relatively easy and phenylalanine tolerance rose remarkably during pregnancy. This course was expected from previous personal experience and from reports in the literature (Hyanek et al 1988). In contrast, dietary management of a PKU patient carrying a PKU-affected fetus was much more difficult and phenylalanine tolerance hardly rose. This suggests that the fetal liver contributes significantly to the metabolism of ingested phenylalanine in phenylketonuric women. Pregnancies with affected fetuses occur when the woman’s partner has PKU (as in patient 3). This can also occur when an affected PKU individual becomes pregnant by an unaffected PAH mutation carrier. Phenylalanine tolerance remaining low in the third trimester of pregnancy may then indicate before birth that the fetus has PKU.
In patient 3, phenylalanine blood concentrations repeatedly exceeded the desirable upper limit in spite of a low phenylalanine intake. She was therefore encouraged to stimulate anabolism by increasing her phenylalanine-free caloric intake. This resulted in a high total gain of her body weight during pregnancy, a factor associated with a relatively low likelihood of microcephaly in the offspring of PKU mothers (Matalon et al 2003). Nevertheless, the birth weight of the newborn, expressed as a proportion of maternal weight gain, was only 16%, while this fraction in non-PKU women ranges from 25 (in wealthy countries) to 60% (in poor countries), depending on the gen eral nutritional situation of the respective population (Prentice and Goldberg 2000). The head circumference of the PKU-affected newborn of patient 3 was on the 20th centile, while it was on the 90th and 75th centiles, respectively, in the two unaffected newborns of patients 1 and 2. Whether the fetal head of patient 3 could have grown more with better maternal control remains speculative. In any case, for the prevention of insufficient fetal head growth and associated brain damage, a very strict metabolic control during the third trimester remains of utmost importance in the pregnancy management of women with PKU.
Conclusion
The difference in phenylalanine tolerance between the pregnancies with PKU-affected and non-affected fetuses suggests that the genetic and metabolic status of the fetus influences maternal metabolic control. If a PKU pregnancy remains difficult to manage in the third trimester because the expected increase of phenylalanine tolerance does not take place, the fetus may be affected with PKU.
Abbreviations
- PAH:
-
phenylalanine hydroxylase
- PAH :
-
phenylalanine hydroxylase gene
- PKU:
-
phenylketonuria
References
Bremer HJ, Bührdel P, Burgard P, et al (1997) Therapie von Patienten mit Phenylketonurie. Monatsschr Kinderheilkd 145: 961–962.
Hyanek J, Viletova H, Soukup J, Kobilkova J, Kubik M, Kunova V (1988) Changes in phenylalanine tolerance while monitoring the dietetic treatment of pregnant women suffering from hyperphenylalaninaemia. J Inherit Metab Dis 11: 427–428. doi:10.1007/BF01800434.
Levy HL (2003) Historical background for the maternal PKU syndrome. Pediatrics 112: 1516–1518.
Levy HL, Guldberg P, Guttler F, et al (2001) Congenital heart disease in maternal phenylketonuria: report from the maternal PKU collaborative study. Pediatr Res 49: 636–642. doi:10.1203/00006450-200105000-00005.
Maillot F, Cook P, Lilburn M, Lee PJ (2007) A practical approach to maternal phenylketonuria management. J Inherit Metab Dis 30: 198–201. doi:10.1007/s10545-007-0436-y.
Matalon KM, Acosta PB, Azen C (2003) Role of nutrition in pregnancy with phenylketonuria and birth defects. Pediatrics 112: 1534–1536.
Prentice AM, Goldberg GR (2000) Energy adaptations in human pregnancy: limits and long-term consequences. Am J Clin Nutr 71: 1226S–1232S.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: John Walter
Competing interests: None declared
References to electronic databases: Phenylketonuria: OMIM 261600.
Rights and permissions
About this article
Cite this article
Kohlschütter, B., Ellerbrok, M., Merkel, M. et al. Phenylalanine tolerance in three phenylketonuric women pregnant with fetuses of different genetic PKU status. J Inherit Metab Dis 32 (Suppl 1), 1–4 (2009). https://doi.org/10.1007/s10545-008-0910-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-008-0910-1