
Abstract
A simple microfluidic immunoassay card was developed based on polystyrene (PS) substrate for the detection of horse IgG, an inexpensive model analyte using fluorescence microscope. The primary antibody was captured onto the PS based on covalent bonding via a self-assembled monolayer (SAM) of thiol to pattern the surface chemistry on a gold-coated PS. The immunosensor chip layers were fabricated from sheets by CO2 laser ablation. The functionalized PS surfaces after each step were characterized by contact angle measurement, X-ray photoelectron spectroscopy (XPS), and atomic force microscopy (AFM). After the antibody–antigen interaction as a sandwich immunoassay with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody, the intensity of fluorescence was measured on-chip to determine the concentration of the target analyte. The present immunosensor chip showed a linear response range for horse IgG between 1 μg/ml and 80 μg/ml (r = 0.971, n = 3). The detection limit was found to be 0.71 μg/ml. The developed microfluidic system can be extended for various applications including medical diagnostics, microarray detection and observing protein–protein interactions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The treatment during acute care using an outside laboratory requires more time from sampling to receiving results, which limits the use of these results for clinical decision making. Point-of-care testing (POCT) systems can overcome the problem and facilitate treatment for patient by providing laboratory results fast and efficiently at the patient’s bedside (St-Louis 2000; Altieri and Camarca 2001; Lode 2005; Yager et al. 2008). Thus, it is highly desirable to develop a low-cost and portable device that will enable the determination of the relative concentrations of the biomarkers of interest for POCT.
The immunoassay is one the most important analytical methods for clinical diagnosis and biochemical studies because of its extremely high specificity (Chan et al. 2003). Of the various solid-phase immunoassay formats in current use, the sandwich immunoassay typically carried out in polystyrene microtiter plates is superior to other types of heterogeneous solid phase immunoassays with respect to sensitivity, specificity and kinetics (Diamandis and Christopoulos 1996). However, this method requires large volumes of reagents and long incubation times due to the high volume-to-surface ratio. The microfluidic devices permit one to address these drawbacks specifically to perform sandwich immunoassay. Based on the results of the studies conducted to date, a microfluidic immunoassay is attractive because it improves the analytical performance by reducing sample and reagent consumption, power consumption and cost. In addition, the surface-to-volume ratio of the microfluidic device can enhance mass transport, resulting in shorter assay times, increased detection sensitivity, and integrating multiple processes in a single device compare to the conventional analytical methods (Becker and Locascio 2002; Erickson and Li 2004).
To date, most of the microfluidic immunoassay systems have been fabricated using silicon, metal, and glass as the substrate material (Bange et al. 2005). However, machining these materials and fabrication is often a time-consuming and expensive process that presents a number of problematic issues that may ultimately hinder their widespread use in commercial applications. On the other hand, polymers are low cost alternative substrate materials for biochip and/or biomedical applications owing to a favorable combination of advantageous properties, such as, mass production, ease of machining and shaping (Mathieu et al. 2003; Bai et al. 2006). The potential of polymers and plastic as substrate materials in microfluidic applications has been reviewed by A. de Mello (de Mello 2002). Of the available polymers, polystyrene (PS) has been widely employed in many industrial and medical fields because of its low cost and high workability. PS is usually utilized in the domain of diagnostics due to its optical transparency, good mechanical properties, durability, and non-toxicity. However, it also presents several general disadvantages—poor chemical resistance, difficulty in controlling surface chemistry, background fluorescence and hydrophobicity—that pose a problem with its applications. Surface modification to improve wettability, adhesion, biocompatibility and topography is thus necessary. A non treated PS surface is by nature hydrophobic, however, it can be easily modified by ion beam treatment, plasma treatment, UV/ozone treatment and graft polymerization to alter the chemistry of surface.
Furthermore, optical biosensors represent one of the major families of biosensors which have been exploited for bioassay applications due to some advantages, such as sensitivity, simplicity and immunity to electromagnetic wave interferences (Ligler and Rawe-Taitt 2002). These sensors have extended the limit of application of the spectrophotometric methods in analytical chemistry particularly for miniaturized systems. Of the variety of optical techniques that have been successfully demonstrated in microfabricated structures, fluorescence is a popular technique as it provides powerful detection tools, increasing sensitivity and gives the possibility of imaging sites on a restricted area for detection of on-chip reaction (Chabinyc et al. 2001; Schilling et al. 2002).
Our laboratory is developing tools for point-of-care instrumentation for monitoring the biomakers in small blood samples using fluorescence technique. One aim is to fabricate polymeric devices that contain primary antibody covalently immobilized onto PS substrates (Darain et al. 2008). A PS support covalently attached with hapten has been reported by Kaur et al. for application in enzyme-linked immunosorbent assay via silanization (Kaur et al. 2004). However, extensive research on surface modification of PS by covalent bonding is still lacking. Therefore, the main issue addressed in this preliminary work is: the development of strategy for simple and highly efficient in-channel immobilization of antibody onto PS for plastic based microfluidic card; sandwich immunoassay based on antibody immobilized card for the detection of horse IgG as a model analyte.
2 Experimental
2.1 Materials and reagents
16-mercapto-hexadeconic acid, 90% (16-MHA), N-hydroxy succinimide (NHS) ester, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, >98%) were purchased from Aldrich, USA. Bovine serum albumin (BSA), horse IgG, it’s primary antibody (anti-hIgG) and secondary antibody conjugated with FITC (anti-hIgG-FITC) were purchased from Sigma, USA. Phosphate buffered saline (PBS, pH 7.4) was used as working medium (Sigma). Unless otherwise stated, all chemicals used in this study were of analytical grade. The deionized water having 18 MΩcm resistivity was used in making all aqueous solutions. The polystyrene (PS), Mylar, and polymethylmethylacrylate (PMMA) sheets were purchased from Professional Plastics, USA.
2.2 Apparatus
The individual layers of the microfluidic devices were fabricated using a CO2 laser cutting tool (Epilog Laser, Legend 24TT, USA). Contact angle measurements were performed using a FTA200 Dynamic Contact Angle Analyzer (USA) combined with a program, FTA 32 utilizing sessile drop method. Contact angle values were calculated using the software provided by the manufacturer. The AFM measurements were performed using a Nanoscope AFM (dimension 3100 scanning probe microscope, Digital Instruments, USA) at tapping mode. XPS analysis was performed using a Kratos AXIS spectrometer (Kratos Analytical Ltd. UK) with monochromatic Al Kα (1486.71 eV) X-ray radiation. The fluorescence signal was detected by using a Zeiss upright epi-fluorescence microscope (Axioskop 2 mot plus, Germany). A mercury arc lamp was used as the light source with the appropriate set of filters for FITC excitation and emission wavelengths. A Zeiss 12-bit cooled CCD camera (AxioCam MRc5, Germany) operated with Auxiovision software was used to acquire images of the channels at an exposure time of 2 s.
2.3 Device fabrication, assembly, and microfluidic system
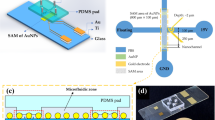
Before fabricating, device design, width (0.2 mm) and length (6.2 mm) of the channels and individual layers were drawn using a computer-aided design (CAD) program, and then transferred to the computer-controlled CO2 laser cutting machine. The channel layout is shown in Fig. 1. The device consisted of 3 layers. Layer 1, was cut from 1 mm thick sheets of PS. Layer 2, is a Mylar channel layer (0.05 mm thick with adhesive layer on both sides), which is applied to the surface of the PS slide and Layer 3, the channel-capping layer of optically clear 100 μm thick PMMA is applied next, forming the top of the immunoassay channel. The protective layers over the adhesive were removed and the laminates are assembled into a composite device (Fig. 1. Top view). The microchip was incorporated in the flow system via a special in-house designed manifold made of PDMS with the inlet and outlet holding O-rings into holes in the top cover (Fig. 1. Over view). The chip was sandwiched between top and bottom parts of the manifold. The two parts are sealed with screws and nuts. A basic microfluidics set-up was used, consisting of a computer-controlled syringe pumps (Kloehn Ltd., USA), with 50 μL syringes and a six-port injection loading valve for sample loading connected by PEEK™ (polyetheretherketone) tubing (1/16 × 0.02 × 5 ft) purchased from Upchurch Scientific, USA. Channels were thoroughly rinsed with buffer prior to each run.

Microfluidic immunoassay device fabrication (not in scale): 1 Au-PS substrate with SAM of 16-MHA, 2 mylar channel layer with adhesive on both sides, 3 optically clear PMMA as channel capping layer, 4 top view of the card, 5 an overview of the device with O-ring to hold connecting tubing in place, 6 Photograph of a device
2.4 Antibody immobilization on PS
PS slides were cut into pieces (2.5 cm × 7.5 cm), first cleaned with freshly prepared piranha solution (1: 3 H2O2—conc. H2SO4) for 12 h, then rinsed with deionized water and dried with N2. The schematic of modification protocol is presented in Fig. 2. The cleaned PS substrates were coated with gold by sputtering. The metal thickness was ~20 nm. The gold coated PS slides (PS-Au) were cleaned using the same cleaning treatment with piranha solution. The 16-MHA self-assembled monolayer (SAM) was prepared by immersing the PS-Au with 1 mM 16-MHA in ethanol for 6 h at room temperature. The substrates were then rinsed with ethanol in order to remove non-bonded thiols and subsequent drying with a N2 stream. Then the SAM modified PS-Au was utilized to make the chip (layer 1). Subsequently, a mixture of 0.4 mM EDC and 0.1 mM NHS was introduced in to the microchannels to convert the terminal carboxylic groups to an active NHS ester. After 25 min the channels were rinsed three times with PBS, pH 7.4. Then a solution of 2 μg/ml of anti-hIgG was flushed through to the channels at 100 nL/s and incubated for 20 min at room temperature (at stopped-flow condition). The excess anti-hIgG was removed by rinsing the channels with PBS for three times. Finally, 1% BSA was flashed through the channels as a blocking reagent followed by rinsing three times with PBS.

Modification steps of PS surface: Here, (a) cleaned PS substrate, (b) PS substrate after Au sputtering, (c) SAM of 16-MHA on Au-PS, (d) after reaction with cross-linker, and (e) after incubation with primary antibody anti-hIgG
2.5 Detection principle and data processing
The anti-IgG-modified chip was placed on the stage (under the objective) of an upright epi-fluorescence microscope with all necessary inlet and outlet tubing and syringe pump. The hIgG was flushed through the microchannels and then rinsed with PBS. Subsequently, anti-hIgG-FITC (2 μg/ml) was flushed through the microchannels. The flow rate of analyte and detection antibody solution was 100 nL/s. The microchannels were thoroughly rinsed with PBS for three times after 5 min between each step at 1 μL/s. The detection of hIgG was carried out in terms of bound FITC in the microchannels under stopped-flow condition. In a negative control experiment, all steps were identical, except that the antigen hIgG incubation step was omitted. The intensity of fluorescence over a selected region of each channel was analyzed with ImageJ (software for image processing and analysis in Java, NIH, USA). The fluorescence intensity in each channel was then calculated by subtracting the intensity obtained for the negative control.
3 Results and discussion
3.1 Antibody binding onto polystyrene and microfluidic system
Among the fabrication techniques that can be used to fabricate lab-on-a-chip devices, the direct write CO2-laser cutting is advantageous because it is very flexible, simple, desktop computer controlled, and fast (Hawkins and Yager 2003). In our study, a simple microfluidic device was constructed according to the schematic shown in Fig. 1. Although polystyrene microbeads have been reported as the solid support for the capture antibody delivered to the channel, despite their advantages, beads have been limited in microfluidics due to their potential to be adsorbed into the channels (Bange et al. 2005). Therefore, it would be advantageous if the PS surfaces can be directly modified chemically with desired antibody. Furthermore, various commonly available surface modification methods have been developed for coupling antibody on to it. For example, antibody immobilization on to a silicon nitride (Diao et al. 2005), a PMMA chip (Wang et al. 2008), a PDMS device (Eteshola and Leckband 2001), a controlled-pore glass (Fernandez et al. 2003), a PMMA microfluidic device (Bai et al. 2006), etc. have been previously reported.
It is well known that the self-assembled monolayers (SAM) approach allows the control of molecular ordering and it is a potentially useful technique for the construction of future advanced materials and devices. Among the techniques of self assembling, gold thiol bonding has been widely used as the formation of SAM occurs due to the strong and spontaneous adsorption between sulfur atoms and a gold surface. This approach of surface modification has been previously utilized onto a Au-coated glass (Mauriz et al. 2006), a Au electrode (Tlili et al. 2006), a piezoelectric crystal (Su et al. 1999), etc. Thus, in the present study, the PS substrate was coated with gold and then SAM of 16-MHA was obtained. Figure 2 shows the protocol of anti-hIgG immobilization on the Au-PS surface. As can be seen from Fig. 2, the hydrophilic –COOH head groups exposed on the surface on the monolayer are available for cross-linking.
On the other hand, non specific protein adsorption to the plastic surfaces is a well known problem (Rebeski et al. 1999). Therefore, prior to the construction of the microfluidic device, the Au-PS was firstly functionalized with SAM of 16-MHA (Fig. 1, layer 1) in order to passivate the surface. Once all the layers of a device were assembled (Fig. 1. Overview), a cross-linker is introduced into the channel interior to achieve the covalent linkage of the desired antibody to the SAM-Au-PS by formation of amide bond between carboxylic acid groups of 16-MHA and amine groups provided by antibody.
3.2 Contact angle measurement of PS surfaces
The measurement of water contact angle is widely used to provide information about surface of materials by revealing the hydrophobic/hydrophilic character of the surface. The drop image was captured by a video camera in room atmosphere; an image analysis software was used to calculate the contact angle from the shape of the drop. In Table 1, listed are the measured contact angles for various PS surfaces after each treatment. Each value reported here is the average of at least five separate drops of water on a given substrate. While the plain cleaned PS slide shows a rather hydrophobic behavior, the contact angle decreases during the treatment, as can be seen from the table. The Au-PS surface is hydrophobic with a contact angle value of ~60, which is lower than PS without Au coating. After thiol deposition the contact angle decreases indicating a more hydrophilic surface (good wettability) which can be attributed to the covalent bonding with such acid thiol. Other groups performed a similar experiment to ours. For example, Tlili et al reported an 11-Mercaptoundecanoic acid self-assembled monolayer on to a gold electrode (Tlili et al. 2006). Our results of the contact angle of measurement for SAM on Au-PS are consistent with that previously observed by Tlili et al. Hence, the formation of the SAM seems to introduce groups to the surface which increase the surface wettability. After the antibody immobilization the contact angle was found to further increase which is also consistent with existing works for anti-rabbit IgG immobilization on to a gold surface via SAM of alkanethiols (Zou et al. 2007). Therefore, these results would not happen if Au-PS surface do not undergo functionalization with first 16-MHA and then with antibody.
3.3 XPS characterization of PS surfaces
XPS technique has already been used to analyze the chemical composition of the material surfaces before and after chemical modifications to evaluate the nature of the chemical bonding occurred on the surface. For this study, we systematically prepared three sets of experiment: blank Au-PS (A), after SAM formation on Au-PS (B), and after anti-hIgG immobilization on SAM-Au-PS (C) as shown in Fig. 3. The XPS survey scans detected gold, carbon, and oxygen in all PS films analyzed. For the Au-PS surface (Fig. 3(a)), C 1 s peak is observed at 285.2 eV as a main constituent of the PS itself, whereas the peak at 532.2 eV is also appeared probably due to the oxygen contamination in bare Au-PS surface. The peak intensity of O 1 s and C 1 s for a blank Au-PS increases after formation of SAM onto it as shown in Fig. 3(b). Moreover, S 2p peak was observed at 163 eV for a SAM-Au-PS while no S 2p peak is observed in the survey spectra of blank Au-PS confirming that Au-S bond is formed. This binding energy is characteristic to the chemical state of the S 2p3/2 in the gold–thiol bond, very consistent with previously published result (Johnson and Levicky 2004). Thus, the above result indicates that 16-MHA was chemisorbed on Au-PS through Au–S bonding and so the –COOH groups are exposed at the surface side in the SAMs (Nakamura et al. 2006). Finally, a strong N 1 s signal at 400.5 eV is observed in survey spectra for the anti-hIgG immobilized SAM-Au-PS as shown in Fig. 4(c). The N 1 s core level spectra is fitted by one component, which is typical binding energy of the amide nitrogen atoms (HN–C=O) coming from the peptide function of antibody (Beamson and Briggs 1992). The observations are in agreement with that of other reported researches, such as an 11-Mercaptoundecanoic acid SAM functionalized with enzyme (Cecchet et al. 2006). Therefore, XPS systematic study clearly shows that the anti-hIgG was successfully coupled on the Au-PS surface via SAM of 16-MHA.

Survey XPS spectra of the gold coated PS surfaces: (a) blank Au-PS, (b). after formation of SAM of 16-MHA on Au-PS, and (c). after immobilization of anti-hIgG on SAM-Au-PS

AFM topographic and 3-D images of various PS surfaces: Both 2- and 3-D images are displayed to show the surface morphology and roughness. (a) blank Au-PS, (b) after formation of SAM on Au-PS, and (c) after immobilization of anti-hIgG on SAM-Au-PS
3.4 AFM characterization of PS surfaces
The topographic features of the PS surfaces before and after modification are revealed by the respective AFM images. The present study has considered three samples for determining the surface morphologies as well as to obtain information on surface modification. Figure 4 represents the topographic and 3-D images (scan area of 1 μm × 1 μm) of the surface of bare Au-PS surface (a), SAM modified Au-PS surface (b), and anti-hIgG immobilized SAM-Au-PS surface (c), respectively. The gold coating on PS produced a relatively homogenous and flat surface. Importantly, the mean roughness, R a for Au-PS is 1.5 nm, whereas for an untreated cleaned PS, the R a is 6.7 nm. It can also be seen from Fig. 3(a) that the Au-PS has smooth surface and no crack or no pore can be observed. Figure 3(b) shows the image for a SAM-Au-PS is obviously different from that of blank Au-PS and the surface becomes rougher. The R a value increases to 2.2 nm compare to that of blank Au-PS. Thus the formation of SAM on to Au-PS has been accomplished and the morphology has been changed. The AFM image of anti-IgG immobilized onto SAM-Au-PS (Fig. 4(c)) also shows a very different morphology from that obtained for SAM-Au-PS and roughness also increases to R a is 5.5 nm. As can be seen from Table 1, each subsequent surface modification increases the apparent surface roughness of Au-PS. Upon immobilization of anti-hIgG onto the SAM-Au-PS, uniform globular structures were clearly visible on the surface. The observation is in agreement with previous report by other researchers for anti-rabbit IgG immobilized on to a poly (vinyl alcohol)-modified poly(dimethylsiloxane) (Yu et al. 2007). Yu et al. also reported that the amount of protein adsorbed nonspecifically on a PDMS surface was nonuniform compared to covalently bound protein. In addition, the nonspecific adsorption of protein to a surface might be prevented by a SAM (Hillebrandt and Tanaka 2001). Therefore, these results of AFM images support the conclusion that there was successful covalent attachment of anti-hIgG on to the Au-PS surface via the SAM of 16-MHA which could be utilized in biosensing systems.
3.5 On-chip fluorescence detection of hIgG
After assembling the microchannels containing primary antibody anti-hIgG and blocking with 1% BSA, various concentrations of hIgG were injected into the sample loading valve and pumped through the microchannels by the syringe pump, followed by rinsing with PBS and incubation with detection antibody anti-hIgG conjugated with FITC. After that the unbound anti-hIgG-FITC were rinsed out with PBS. The hIgG was bound between two antibodies, a surface-immobilized ‘capture’ anti-hIgG and a ‘detection’ anti-hIgG-FITC antibody. The amount of fluorescence signal is dependent on the amount of anti-hIgG-FITC bound on the top layer of the immunoassay sandwich—and thus the concentration of hIgG bound in the layer before. Fluorescence measurements of bound anti-hIgG-FITC were carried out at the completion of the immunoassay and CCD camera images of the microscope were quantified for intensity. The images taken after the immunoassay are shown in Fig. 5(a). During the measurement the chip was fixed on the microscope stage using a special holder and thus images were captured at the same region at for each microchannel. The variation of fluorescence intensity in the same microchannel is within 5%. The used pumping rate dramatically decreases the adsorption time and achieves a saturation state in a shorter period of time. Under optimum conditions, quantification of the fluorescence signal was done and calibration curve is plotted as shown in Fig. 5(b). As can be seen from Fig. 5(b), the intensity is proportional to the hIgG concentration in the test sample. The intensity starts to become saturated as the concentration of hIgG approaches 80 μg/ml. The linear range of quantification of this model analyte hIgG was found from 1 μg/ml to 80 μg/ml. This linear dependency of the hIgG concentration yields the regression equation of (intensity) = (219.49 ± 7.4) + (6.98 ± 0.7) [hIgG], μg/ml with correlation coefficient of 0.971 (SD: ±56.39, n = 3). The lower detection limit calculated based on the signal to noise ratio of 3 is 0.71 (±0.08) μg/ml. The value of detection limit obtained from our system for horse IgG as a model analyte is comparable to that of some reported microfluidic systems. The present detection limit is found to be lower than that of a multiplexed microfluidic immunoassay system for human IgG (1 μg/ml) based on avidin–biotin bridge for antibody immobilization on to poly(dimethylsiloxane) by surface plasmon resonance imaging (Dong et al. 2008) and a microfluidic system integrated with two-dimensional surface plasmon resonance phase imaging system for rabbit IgG (0.1 μg/ml) based on covalently immobilized antibody on to a SAM on gold (Lee et al. 2007). Furthermore, the present detection limit is found to be higher than those of previously reported microfluidic systems, for instance, the obtained detection limit is higher than that of a polydimethylsiloxane microfluidic immunoassay system for sheep IgG (15 ng/ml) might be due to the fact that they used different detection strategies where enzyme tracers were involved (Eteshola and Leckband 2001) and that of a screen-printed microfluidic electrochemical immunoassay for mouse IgG (10 ng/ml) might be due to the different set up and detection strategy (Dong et al. 2007). However, we believe that the present system can find potential application in the field of biomedical research because the modification of PS surface is simple and the device fabrication is fast. Moreover, the stability of the chip as sensor is also good might be due to covalent attachment of primary antibody onto the chip.

(a) Fluorescence microscopic images of the channels for negative control (left) and with antigen hIgG (right). Scale bars are 200 μm. (b) Calibration plot for the sandwich immunoassay in a microfluidic chip. The graph indicates the normalized fluorescence intensity as a function of the concentration of hIgG in PBS. The averages and error bars were calculated from three independent measurements
The assessment of selectivity of the present immunosensor chip was performed using goat IgG. It can be noted that no change in the fluorescence intensity was observed for goat IgG compared to that of horse IgG and was similar to that of a negative control. Therefore, this result indicates that the antigen–antibody interaction was specific as expected. The chip response precision (in terms of relative standard deviation) at 10 μg/ml of hIgG is 7.1%. The storage ability of the sensor chip was examined when it was stored at 4°C after preparation and the immunoassay was performed on each seven days. It was found that the magnitude of fluorescence intensity remained more or less constant for 56 days and then gradually decreased.
In the present study, under the selected conditions described above, the total immunoassay time required for the hIgG measurements (including injection, incubation and detection steps) was found to be approximately 25 min while hours or days is required in traditional enzyme-linked immunosorbent assay (ELISA). In this assay the total consumption of each reagent was 50 μl which is much less than that of the traditional assay in microtiter plates where typically about 100 μl of each reagent is required. The reagent consumption can be further reduced by using short length of tubing. Therefore, the present results, however, demonstrate that due to low cost and easy-handling process the developed chip has the potential in clinical diagnosis.
4 Conclusions
In conclusion, a microfluidic immunosensor chip was developed based on an inexpensive polymer, polystyrene with on-chip fluorescence imaging for the detection of horse IgG, as a model analyte. The modification of PS is simple and the fabrication of microfluidic card is fast and inexpensive manufacturing process is necessary. The system provided a much shorter analytical time than that of the conventional method. Also the consumption of the reagents was significantly reduced. Moreover, the assay with the microfluidic chip is advantageous because of simple operations, flow rate control with syringe pumps compare to troublesome liquid handling procedures in the conventional assay. The preliminary results indicate that the system described herein could be further miniaturized potentially to develop one robust plastic based disposable immunosensor chip and could be extended to an integrated system (with an excitation source, a light guide and a fluorescence detector) for point-of-care bioassay system, in future.
References
M.F. Altieri, M. Camarca, Point of care testing Clin. Pediatr. Emerg. Med 4, 275–279 (2001). doi:10.1016/S1522-8401(01)90036-9
Y. Bai, C.G. Koh, M. Boreman, Y.-J. Juang, I.-C. Tang, L.J. Lee, S.-T. Yang, Surface modification for enhancing antibody binding on polymer based microfluidic device for enzyme-linked immunosorbent assay Langmuir 22, 9458–9467 (2006). doi:10.1021/la061123l
A. Bange, H.B. Halsall, W.R. Heineman, Microfluidic immunosensor system Biosens. Bioelectron 20, 2488–2503 (2005). doi:10.1016/j.bios.2004.10.016
G. Beamson, D. Briggs, High Resolution XPS of Organic Polymers The Scienta ESCA Database (Wiley, Chichester, U.K., 1992)
H. Becker, L.E. Locascio, Polymer microfluidic devices Talanta 56(2), 267–287 (2002). doi:10.1016/S0039-9140(01)00594-X
F. Cecchet, M. Marcaccio, M. Margotti, F. Paolucci, S. Rapino, P. Rudolf, Redox mediation at 11-mercaptoundecanoic acid self-assembled monolayers on gold J. Phys. Chem. B 110, 2241–2248 (2006). doi:10.1021/jp054290n
M.L. Chabinyc, D.T. Chiu, J.C. McDonald, A.D. Stroock, An integrated fluorescence detection system in poly(dimethylsiloxane) for microfluidic applications Anal. Chem. 73(18), 4491–4498 (2001). doi:10.1021/ac010423z
C.P.Y. Chan, W.S. Cheng, J.F.C. Glatz, D.V.D. Voort, J.E. Sanderson, A. Hempel, M. Lehmann, I. Renneberg, R. Rennerberg, Early diagnosis of acute myocardial infarction using immunosensors and immunotests Anal. Lett. 36(9), 1987–2004 (2003). doi:10.1081/AL-120023625
F. Darain, P. Yager, K.L. Gan, S.C. Tjin, On-chip detection of myoglobin based on fluorescence. Biosens. Bioelectron. (2008). doi:10.1016/j.bios.2008.09.004
A. de Mello, Plastic fantastic? Lab Chip 2, 31N–36N (2002)
E.P. Diamandis, T.K. Christopoulos (eds.), Immunoassay (Academic, New York, 1996)
J. Diao, D. Ren, J.R. Engstrom, K.L. Lee, A surface modification strategy on silicon nitride for developing biosensors Anal. Biochem. 343, 322–328 (2005)
H. Dong, C.-M. Li, Y.-F. Zhang, X.-D. Cao, Y. Gan, Screen-printed microfluidic device for electrochemical immunoassay Lab Chip 7, 1752–1758 (2007)
Y. Dong, T. Wilkop, D. Xu, Z. Wang, Q. Cheng, Microchannel chips for the multiplexed analysis of human immunoglobulin G–antibody interactions by surface plasmon resonance imaging Anal. Bioanal. Chem. 390, 1575–1583 (2008)
D. Erickson, D. Li, Integrated microfluidic devices Anal. Chim. Acta 507(1), 11–26 (2004)
E. Eteshola, D. Leckband, Development and characterization of an ELISA assay in PDMS microfluidic channels Sens. Actuat. B 72, 129–133 (2001)
P. Fernandez, J.S. Durand, C. Perez-Conde, G. Paniagua, Permanently oriented anibody immobilization for digoxin determination with a flow-through fluoroimmunosensor Anal. Bioanal. Chem. 375, 1020–1023 (2003)
K.R. Hawkins, P. Yager, Nonlinear decrease of background fluorescence in polymer thin-films—a survey of materials and how they can complicate fluorescence detection in μTAS Lab Chip 3, 248–252 (2003)
H. Hillebrandt, M. Tanaka, Electrochemical characterization of self-assembled alkylsiloxane monolayers on indium–tin oxide (ITO) semiconductor electrode J. Phys. Chem. B 105, 4270–4276 (2001)
P.A. Johnson, R. Levicky, X-ray photoelectron spectroscopy and differential capacitance study of thiol-functional polysiloxane films on gold supports Langmuir 20(22), 9621–9627 (2004)
J. Kaur, K.V. Singh, M. Raje, G.C. Varshney, C.R. Suri, Strategies for direct attachment of hapten to a polystyrene support for application in enzyme-linked immunosorbent assay (ELISA) Anal. Chim. Acta 506, 133–135 (2004)
K.-H. Lee, Y.-D. Su, S.-J. Chen, F.-G. Tseng, G.-B. Lee, Microfluidic systems integrated with two-dimensional surface plasmon resonance phase imaging systems for microarray immunoassay Biosens. Bioelectron. 23, 466–472 (2007)
F.S. Ligler, C.A. Rawe-Taitt (eds.), Optical Biosensor: Present and Future (Elsevier, Amsterdam, 2002)
P.V. Lode, Point-of-care immunotesting: approaching the analytical performance of central laboratory methods Clin. Biochem. 38(7), 591–606 (2005)
H.J. Mathieu, Y. Chevolot, L. Ruiz-Taylor, D. Leonard, Engineering and characterization of polymer surfaces for biomedical applications Adv. Polym. Sci. 162, 1–34 (2003)
E. Mauriz, A. Calle, A. Abad, A. Montoya, A. Hildebrandt, D. Barcelo, L.M. Lechuga, Determination of carbaryl in natural water samples by a surface plasmon resonance flow-through immunosensor Biosens. Bioelectron. 21, 2129–2136 (2006)
F. Nakamura, E. Ito, T. Hayashi, M. Hara, Fabrication of COOH-terminated self-assembled monolayers for DNA sensors Colloids Surf. A: Physicochem. Eng. Aspects 284–285, 495–498 (2006)
D.E. Rebeski, E.M. Winger, Y.K. Shin, M. Lehenta, M.M. Robinson, R. Verecka, J.R. Crowther, Identification of unacceptable background caused by non-specific protein adsorption to the plastic surface of 96-well immunoassay plates using a standardized enzyme-linked immunosorbant assay procedure J. Immunol. Methods 226, 85–92 (1999)
E.A. Schilling, A.E. Kamholz, P. Yager, Cell lysis and protein extraction in a microfluidic device with detection by a fluorogenic enzyme assay Anal. Chem. 74(8), 1798–1804 (2002)
P. St-Louis, Status of point-of-care testing: promise, realities, and possibilities Clin. Biochem. 33(6), 427–440 (2000)
X.D. Su, F.T. Chew, S.F.Y. Li, Self-assembled monolayer based piezoelectric crystal immunosensor for the quantification of total human immunoglobulin E Anal. Biochem. 273, 66–72 (1999)
A. Tlili, A. Abdelghani, S. Ameur, N. Jaffrezic-Renault, Impedance spectroscopy and affinity measurement of specific antibody–antigen interaction Mater. Sci. Eng. C 26, 546–550 (2006)
H. Wang, S. Meng, K. Guo, Y. Liu, P. Yang, W.Z.B. Liu, Microfluidic immunosensor based on stable antibody-patterned surface in PMMA microchip Electrochem. Commun. 10(3), 447–450 (2008)
P. Yager, Gonzalo, J. Domingo, J. Gerdes, Point-of-care diagnostics for global health Annu. Rev. Biomed. Eng. 10, 107–144 (2008)
L. Yu, C.M. Li, Q. Zhou, J.H.T. Luong, Poly(vinyl alcohol) functionalized poly(dimethylsiloxane) solid surface for immunoassay Bioconjugate Chem 18(2), 281–284 (2007)
Z. Zou, J. Kai, M.J. Rust, J. Han, C.H. Ahn, Functionalized nano interdigitated electrodes arrays on polymer with integrated microfluidics for direct bio-affinity sensing using impedimetric measurement Sens. Actuat. A: Phys. 136(2), 518–526 (2007)
Acknowledgements
This work was supported by Biomedical Engineering Research Council of Singapore under the Singapore-University of Washington Alliance program. F. Darain would like to thank Ms. Fang Xiaoqin from School of Electrical and Electronic Engineering for her help to make the design to cut the layers for microfluidic devices.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Darain, F., Gan, K.L. & Tjin, S.C. Antibody immobilization on to polystyrene substrate—on-chip immunoassay for horse IgG based on fluorescence. Biomed Microdevices 11, 653–661 (2009). https://doi.org/10.1007/s10544-008-9275-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10544-008-9275-3