
Abstract
We have developed a simple microfluidic device for generating stable concentration gradients in 2D and 3D environments. The device, termed the Ladder Chamber, uses a two-compartment diffusion system to generate steady state gradients across flow-free channels that connect the source and sink channels. To demonstrate the utility of the Ladder Chamber for cell migration, neutrophil chemotaxis was successfully observed in soluble chemoattractant (IL-8) gradient. The Ladder Chamber’s simple design and experimental implementation make it an attractive approach for investigating cell migration and other biological experiments in well-defined gradients in 2D surfaces as well as in 3D gels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Chemotaxis, directed cell migration in concentration gradients of soluble factors, has been a focus of intense research for the past three decades, and is implicated in a number of biological processes such as inflammation (Yang et al. 1999), wound healing (Maheshwari et al. 1999), and cancer metastasis (Condeelis et al. 2001; Kassis et al. 2001; Wells et al. 2002). A variety of assays have been developed for chemotaxis, including the Boyden chamber (Transwell assay; Boyden 1962), under-agarose assay (Nelson et al. 1975), Zigmond chamber (Zigmond 1977), Dunn chamber (Zicha et al. 1991), and micropipette assay (Gerisch and Keller 1981). The Boyden chamber measures chemotaxis as the number of cells that migrate across a filter between two compartments containing soluble factors (Boyden 1962). In the under-agarose assay, cells migrate on a glass coverslip, underneath an agarose gel, with wells for cells and chemoattractants punched into the agarose (Nelson et al. 1975). The Zigmond and Dunn chambers consist of two wells separated by a bridge, across which a gradient forms by diffusion between the two wells; cells migrate on a glass coverslip placed against the bridge (Zicha et al. 1991; Zigmond 1977). Micropipettes releasing chemoattractants have also been used to generate gradients and observe chemotaxis (Bailly et al. 1998; Gerisch and Keller 1981; Xiao et al. 1997). More recently, an optical chemotaxis assay system was reported, producing precisely defined gradients across microfabricated channels in silicon substrates for the study of neutrophils chemotaxis (Kanegasaki et al. 2003).
While each assay has its own advantages, it is difficult to maintain stable concentration gradients over time (several hours) using conventional assays. This may be inherent in the assay principle itself, as in the case of the under-agarose assay, which relies on radial diffusion in an infinite plain (Lauffenburger and Zigmond 1981). In other cases, gradient instability may result from experimental limitations, such as accumulation/depletion in the reservoirs, as in the case of the Zigmond chamber and the optical chemotaxis chamber. A notable exception is the Dunn Chamber, which allows stable gradients to be produced with its precise construction. Modified micropipette assay systems have also been capable of generating stable gradients (Lohof et al. 1992; Soon et al. 2005; Zheng et al. 1994).
In addition to gradient instability, conventional assays suffer from a number of other limitations. For example, the Boyden chamber, being an endpoint assay, does not allow chemotaxis to be visually observed, and cannot directly distinguish chemotaxis from chemokinesis. The use of a filter as a migration substrate is also the source of various confounding variables, further obscuring the migratory response (see reviews by Wilkinson (1998) and Wells (2000) for details). In the case of the micropipette assay, the main drawback is its low throughput. Though a wealth of information about the dynamics of the migration process can be obtained with this assay, statistical assessments are often difficult and labour-intensive. The assay is thus best suited for qualitative, single-cell studies.
A microfluidics-based method for generating stable concentration gradients with controlled profiles has been developed (Dertinger et al. 2001; Jeon et al. 2000). The approach permits gradients of complex shapes to be produced, maintaining constant gradients for long periods of time, as well as allowing the gradients to be dynamically manipulated during the course of the experiment (Lin et al. 2004a,b). This microfluidic assay has been successfully applied to study the chemotaxis of neutrophils (Jeon et al. 2002; Lin et al. 2004a,b, 2005) and breast cancer cells (Saadi et al. 2006; Wang et al. 2004), as well as the proliferation and differentiation of human neural stem cells (Chung et al. 2005). However, the microfluidic chemotaxis chamber has two main drawbacks. First, since the chamber depends on diffusion across laminar streams to establish gradients, the cells are exposed to constant shear flow, which can influence directional motility (Walker et al. 2005). In addition, most of the autocrine/paracrine factors that are secreted by the cells will be washed away. Although this can be advantageous when teasing out the roles of exogenous factors independent of autocrine and paracrine signaling, it is not desirable in other cases when investigating migration or differentiation in the presence of such signaling.
Second, as in most conventional chemotaxis assays, migration in the microfluidic chemotaxis chamber is limited to two-dimensional (2D) substrates. Two-dimensional cell migration assays address a number of important physiological events, such as the migration of neutrophils on the luminal surface of blood vessels before transmigration into surrounding tissues, and the migration of keratinocytes as they repair the extracellular matrix (ECM) during wound healing (Friedl et al. 1998; Moghe et al. 1995). They do not, however, address many other in vivo processes that involve migration in a three-dimensional (3D) matrix, such as neutrophil chemotaxis after transmigration, and cancer cell invasion (Friedl et al. 1998; Moghe et al. 1995).
Several techniques have been employed to generate concentration gradients of soluble factors in 3D gels, to investigate cell migration in tissues-like environments. One approach is to allow diffusion from a source (Moghe et al. 1995) or between a source and a sink (Haddox et al. 1991, 1994) to establish gradients in 3D gels, but the gradient continuously decays. The addition of a barrier with a small gap at its end, between the source and the gel, serves to physically constrain diffusion and slow down the decay of the gradient (Knapp et al. 1999). Simplicity and added gradient stability make this an attractive approach, but the analysis of migration is more complicated, due to the radial nature of the gradient. The gradients that are generated are also nonlinear. Stable linear gradients require that the source and sink to be regularly replenished to prevent depletion and accumulation (Cao and Shoichet 2001). An alternative approach relies on perfusing the gel with permeable hollow fibers; diffusion across the fiber walls and through the gel produces a constant linear gradient (Fisher et al. 1989). Though elegant, the complicated setup for this technique limited its use. A recent approach utilized micropumps to print gradients of soluble factors onto a gel (Rosoff et al. 2004, 2005). This method provides greater flexibility in generating gradients of various shapes, and has the added capability of producing multiple gradients in parallel for higher throughput. As in most of the previous techniques, however, the resulting gradients are unstable, limiting the use of this assay to large molecules.
This paper presents a simple microfluidic approach to generate stable concentration gradients of soluble factors. Using a two-compartment diffusion chamber (Ladder Chamber), steady state concentration gradients were generated across 2D surfaces and in 3D gels, in the absence of shear flow. Neutrophil chemotaxis was successfully demonstrated in the Ladder Chamber. With its simple design and ease of experimental implementation, the Ladder Chamber is an attractive approach for investigating cell migration, especially in 3D environments.
2 Materials and methods
2.1 Fabrication of Ladder Chamber
Ladder Chambers were fabricated using soft lithography and rapid prototyping (Whitesides et al. 2001). Master molds were fabricated by patterning thick negative SU8 photoresist (MicroChem, Newton, MA) on Si wafers (Silicon, Boise, ID). For 2D microgroove Ladder Chambers, two layers of photoresist were patterned. The microgrooves were defined by patterning a 5 μm layer of SU8-5. A thicker, 100 μm layer of SU8-50 was then patterned to form the main channels. For 3D chambers, all the channels were fabricated in one layer of 300 μm thick SU8-100. Positive replicas with embossed channels were fabricated by molding PDMS (Sylgard 184, Dow Corning, Midland, MI) against the master. The cured PDMS was peeled off, and holes were punched (with sharpened needles or stainless steel tubes) for use as reservoirs or fluidic interconnects. The PDMS was irreversibly bonded to a glass slide upon treating both with an air-plasma (Plasma cleaner Model PDC-001, Harrick Plasma, Ithaca, NY), forming the microfluidic chamber.
2.2 Fabrication of 3D gels
Collagen type I (rat tail, BD Biosciences, MA) was mixed with 5× DMEM and a 10× reconstitution buffer comprised of NaHCO3, HEPES buffer (Invitrogen, Carlsbad, CA), and NaOH, to make a ∼3 mg/mL prepolymer solution at 4°C. Immediately after mixing, the prepolymer solution was injected into the Ladder Chamber at room temperature, filling the entire chamber. Vacuum suction was applied at the outlet for 1–2 s to remove the prepolymer solution from the main channels, filling them with air, while leaving the collagen solution in the bridge channels. The collagen was allowed to polymerize for 10 min at 37°C, and the chamber was filled with PBS. The chamber was placed in a 37°C incubator for 30 min to complete the polymerization process, and was then stored at 4°C until use.
2.3 Generation and characterization of gradients
Gradients were characterized using fluorescence microscopy. PBS was loaded into one inlet reservoir (for the sink channel) and a 100 μM FITC-dextran solution (MW 10 kDa, Sigma-Aldrich) in PBS was loaded into the other (for the source channel). Flow rates of ∼0.6–6.0 μL/min were maintained by withdrawing the fluids from the outlet using a syringe pump. Gradient evolution was monitored with fluorescence time-lapse microscopy at room temperature. Images were acquired at 2 frames/min with a CCD camera (CoolSNAP cf, Photometrics, Tucson, AZ) on an inverted microscope (TE300, Nikon, Meville, NY). Images were analyzed using MetaMorph (Molecular Devices, Sunnyvale, CA). For each frame, the fluorescence intensity profile was measured, background-subtracted, and normalized to the maximum intensity in that frame. For the concentration vs. time plot (Fig. 5(c)), the intensity was normalized as above, and the baseline was subtracted in each plot, to account for the slight increase over time in the baseline of the profile in Fig. 5(b).
The theoretical profile of fluorescence evolution in Fig. 5(c) was obtained from the time-dependent solution of the diffusion equation in one-dimension, with fixed boundary conditions:
where C is the concentration of the diffusing molecule, D is the diffusion coefficient, C 0 is the initial concentration throughout the chamber, C L is the concentration in the left main channel, C R is the concentration in the right channel, L is the length of the bridge channel, x is the position across the bridge channel, and t is time. This solution is equivalent to that given for diffusion in the Zigmond chamber (visual assay) by Lauffenburger and Zigmond (1981), and to that for diffusion in a plane sheet (Crank 1964). Note that in our case, C 0 = C L. D for FITC-dextran (MW 10 kDa) in dilute solutions was estimated from molecular weight (Tanford 1961; 1.33 × 10−6 cm2/s), and n ranged from 1 to 5,000. The theoretical and experimental profiles in Fig. 5(c) were fitted to a logistic function and compared using the Fit-Comparison test in Origin (OriginLab, Northampton, MA).
2.4 Neutrophil migration
Human blood was obtained from healthy volunteers, and neutrophils were isolated by density gradient centrifugation (Robbins Scientific, Sunnyvale, CA; Lin et al. 2004a,b). Isolated neutrophils were resuspended at 107 cells/mL in HBSS (Invitrogen, Carlsbad, CA) and kept at room temperature until use. All experiments were performed within 8 h of isolation. Solutions of IL-8 (Sigma-Aldrich) were prepared in RPMI 1640 medium (Invitrogen) with 0.2% BSA (Sigma-Aldrich). A new chamber was used for each experiment. Chambers were coated with fibronectin (BD Bioscience, MA) at 0.5 mg/mL for 1 h at room temperature and blocked with 0.2% BSA in RPMI 1640 for another hour before use. The microscope stage was enclosed in a temperature-controlled box maintained at 37°C. The Ladder Chamber was placed on the microscope stage and neutrophils were introduced by flowing them from the inlet of the sink channel using a micropipette. The neutrophils attached randomly within the device, most of the cells were attached on the sink channels and smaller number was attached on the bridge channel and source channel. The chemoattractant (solution of IL-8, 50 ng/mL, in RPMI 1640) and media (RPMI 1640) were introduced simultaneously using a multi-pipette into the source and sink channels, respectively. Chemoattractant gradients across the bridge channels were obtained by withdrawing the fluids from the outlet using a syringe pump. Flow rates of ∼0.6 μL/min were used. Time-lapse images were acquired at 10 s intervals after steady state concentration gradient was reached.
3 Results and discussion
3.1 Generation of gradients in microgrooves
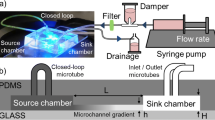
The basic design of the Ladder Chamber consists of two main parallel channels connected by a number of smaller, horizontal bridge channels (Fig. 1). The bridge channels were designed in the form of microgrooves, with dimensions that were significantly smaller (3–10 μm high and 10 μm wide) than the main channels (100 μm high and > 1 mm wide). This design results in high fluidic resistance across the microgrooves compared to the main channels, confining bulk fluid flow in the main channels, and minimizing the likelihood of cross-flow through the microgrooves. Diffusion would thus be the predominant mode of transport across the microgrooves. When a molecular species is introduced in one of the main channel and buffer in the other, molecules diffuse across the microgrooves and generate a gradient. This is equivalent to the diffusion of molecules across a thin film in a source/sink diffusion system. When such a system reaches steady state, a linear concentration is established, the profile of which is given by (Cussler 1997; Crank 1964):
where C is the concentration of the diffusing molecule, C L is the concentration in the left main channel, C R is the concentration in the right channel, L is the length of the bridge channel, and x is the position across the bridge channel. Thus, static source and sink reservoirs were effectively replaced with source and sink microchannels, using constant flow to avoid depletion and build up of molecules.

Design of the Ladder Chamber for generating concentration gradients in microgrooves. a The chamber is fabricated PDMS bonded to glass. Two large parallel channels are connected by horizontal microgrooves. b Phase micrograph of the microgrooves. c Schematic cross-sectional view of the chamber, highlighting the relative dimensions of the microgrooves and main channels, h << H (not drawn to scale)
Figure 2 shows the development of a fluorescence gradient across a 450 μm long microgroove over time. FITC-dextran (100 μM, MW 10 kDa) and PBS were introduced into the top inlets while withdrawing from a common outlet with a syringe pump. The fluorescence intensity of each image was measured and normalized to the standards. (Dertinger et al. 2001; Jeon et al. 2000) Because the height of main channel was much higher (10–30 times) than those of bridge channels the fluorescence signal in the bridge channels was significantly weaker than the main source channel. Thus, higher concentration of FITC-dextran (100 μM) than normally used (10 μM) was used to obtain full scale signal within the bridge channel. This in turn saturated the fluorescence signal in the main channel. When measuring fluorescence intensity across the bridge channel, signal near the source channel (right boundary) were not plotted—this is the reason the intensity is plotted to ∼400 μm and not the whole 450 μm in Fig. 2(b). Based on diffusion calculations (see Section 2), the system is expected to reach 99% equilibrium in 13 min. Experimentally, a linear gradient was established at steady state after 15–18 min in ten separate experiments (Fig. 2(b)), in good agreement with theoretical predictions. Slight deviation between experimental and theoretical values was likely due to variations in channel dimensions, resulting from the SU8 lithography process. Tighter tolerance in microfabrication, achieved with more precise molds (such as micromachined silicon-based molds) may minimize such deviations. Slight variations in initial conditions may also contribute to these deviations.

Development of fluorescence gradients in microgrooves. a Right and left inlets were loaded with FITC-dextran (MW 10 kDa) and PBS, respectively. The microgroove was 450 μm long. Dashed line indicates the location of fluorescence measurement. b Plot of the fluorescence intensity profile across the microgroove as a function of time. A linear steady state gradient was established after 18 min
After reaching steady state, the gradient remained constant after 45 min, and was still stable at 90 min (not shown). It must be emphasized that gradient stability is dependent on the flow, and remains stable as long as the flow is maintained in the main channels. The flow rates and reservoir volumes determine the time window for stable gradients. With typical reservoir sizes (vol = ∼200 μL each reservoir), and a flow rate of 1 μL/min in each channel, the gradients can be maintained for 3 h. The reservoirs can be made larger for longer experiments. The stability of the gradient was aided by the symmetric design of the Ladder Chamber; withdrawal from the outlet induced simultaneous flow from both inlets with equal rates, provided that fluid levels in the inlet reservoirs were balanced.
To demonstrate the use of the Ladder Chamber as a chemotaxis assay, neutrophils were loaded into the chamber, and their migration was observed in IL-8 chemokine gradients. Figure 3 shows time-lapse images of neutrophils migrating in a linear 0–50 ng/mL IL-8 gradient (left to right) inside a microgroove. Cells exhibited highly persistent locomotion in the direction of increasing IL-8 concentration. In this manner, a large number of cells can be observed migrating in a single groove (see supplementary movie), providing large sample sizes and allowing high throughput characterization of cell migration. Although the width of the microgroove in this experiment was small (50 μm), wider designs may be used to allow for movements perpendicular to the direction of the gradient.

Time-lapse images of neutrophils migrating in a microgroove having a 0–50 ng/mL IL-8 gradient. Three cells are observed to traverse the microgroove over the course of 8 min, exhibiting high persistence and high locomotion speeds. Cell #3 exits the field of view after 4 min
A key advantage of the Ladder Chamber is that it allows cell migration experiments in the absence of shear flow. While certain cell types, such as endothelial cells, experience shear force as part of their normal physiology (Nerem et al. 1998), many other cell types in vivo migrate in 3D tissue environments that lack flow. It would be more physiologically relevant to investigate the migration of such cells in flow-free microenvironments. Moreover, the absence of flow makes it possible to study chemotaxis in the presence of autocrine and paracrine molecules, which would otherwise be washed away. Such cell–cell signaling can play an important role in migration, as in the case of macrophages promoting the invasion of breast cancer cells via a paracrine loop (Goswami et al. 2005).
The Ladder Chamber’s compatibility with microscopy is complemented by its simplicity and ease of use. The “ladder” design enables multiple experiments to be performed in parallel; using a number of parallel grooves of varying length, gradients of different steepness can be generated in a single chamber. By increasing the number of inputs in the chamber, it may be possible to generate gradients of different concentrations in a single chamber. The chamber also allows dynamic control of the gradient during the course of the experiment, by simply exchanging the solutions in the inlets.
3.2 Generation of gradients in 3D gels
Generation of stable soluble gradients in 3D gels has been a technical challenge for assays of 3D chemotaxis. The simplest approach, theoretically, relies on diffusion between a source and a sink to generate steady state linear gradients. This, however, assumes that the source and sink are infinite in size compared to the gel. Experimentally, typical dimensions render this assumption invalid, and the resulting depletion and accumulation in the reservoirs induce the gradient profile to change. It has been possible to maintain stability with manual replenishment of the reservoirs (Cao and Shoichet 2001), or with a dual hollow fibre perfusion system (Fisher et al. 1989). Recently, Wu et al. (2006) have reported a novel approach for building complex gradients of diffusible molecules in microfluidic channels. However, a simple and straightforward method for maintaining stable gradients in 3D gels is still lacking.
The Ladder Chamber allows stable gradient to be generated in 3D gels with a fluidic source/sink system. This is made possible by the “ladder” design of the chamber, which allows gels to be selectively localized to the bridge channels between the source and sink streams. We demonstrate this for collagen type I gels. The chamber was first filled with collagen prepolymer solution; vacuum suction was briefly applied to the outlet to remove the prepolymer solution from the inlets and main channels while leaving collagen in the horizontal channels. The collagen was allowed to polymerize, and the channel was then filled with PBS. Even if some residue of collagen is present after the gels are polymerized, the residue remains on the surface of the glass (rather than filling the height of the side channel) and its influence on fluid flow is negligible. The fact that gradient profiles are uniform, and that their evolution experimentally matches that theoretically, indicates that the side flows are well balanced, and that no cross flow occurs when they are filled by gels.
This provides a simple approach for fabricating 3D gels in microfluidic channels without the need for micromolding procedures (Tang et al. 2003). Gel microstructures that are suitable for cell migration studies can thus be readily fabricated. Previous 3D assays used gels that were several mm in size, orders of magnitude larger than typical cell dimensions. The large size of such gels limited the accuracy and reliability of gradient conditions across a given cell diameter. While 200 μm variations between gels are not uncommon in such macro-scale assays (Knapp et al. 1999), the approach reported here allows the fabrication of small 3D gels with higher precision and reproducibility.
The main constraint for gel dimensions in our approach is the ratio of length to width (L:W) of the bridge channels (Fig. 4(a)). Larger L:W ratios (longer, narrower channels) resulted in a higher yield of gels localized in bridge channels, while smaller ratios (shorter, wider channels) increased the possibility of bubbles being trapped within the gels, resulting in a lower yield. We have found the lower limit to be ≈1:1 (Fig. 4(b)), with ratios of 2.5:1 and 5:1 (Fig. 4(c)) yielding more reproducible results. The gel thickness was set to ∼300 μm, similar to that used in studies of 3D cell migration (Friedl et al. 1993). To verify that the entire height of the bridge channel was filled with collagen, we observed the localization of Trypan Blue to the main channels, which showed no evidence of leaks across the gel (data not shown).

Design of the Ladder Chamber for generating concentration gradients in 3D gels. a Schematic of the chamber with gels in the horizontal bridge channels. b, c Phase images of collagen I gels formed within the horizontal bridge channels. The height of the gel was equal to the height of the main channel (∼300 μm). The gels were fabricated by filling the entire chamber with collagen prepolymer solution, followed by brief suction (not shown, see Section 2)
Once the gels were localized in the bridge channels, concentration gradients were established in the gels by placing 0 and 100% FITC-dextran (MW 10 kDa) solutions into the two inlet reservoirs; withdrawal from a common outlet maintained constant flow in the main channels. FITC-dextran diffused between the source and sink channels across the gels, and the gradient developed in a fashion similar to that in the microgrooves (Fig. 5(a)). For 1 mm long gels, linear gradients were established within 1 h (Fig. 5(b)). Gradient evolution in the gel was in close agreement with predictions based on diffusion calculations (F value = 0.95, P = 0.44), using a diffusion coefficient D = 1.33 × 10−6 cm2/s for FITC-dextran (10 kDa) in dilute solutions (Fig. 5(c)). This value of D is consistent with measurements reported by Rosoff et al. (2005), for casein (MW 24 kDa) in 2 mg/mL collagen gels (D for 10 kDa FITC-dextran can be estimated from their measurements to be ∼0.93 × 10−6 cm2/s). This suggests that at the 3 mg/mL collagen concentration, the gels were sufficiently porous to allow FITC-dextran to diffuse freely, as if in solution, without being hindered by the collagen fibres.

Development of fluorescence gradients in collagen I gels. a FITC-dextran (MW 10 kDa), flowing in the right channel, diffused through the collagen gels. Gels were 1 mm long, 200 μm wide, and 300 μm thick. Fluorescence intensity was measured over the length of the middle bridge channel, and averaged over a width of 30 pixels (horizontal dashed lines). b Intensity profiles across the gel during gradient development. c Evolution of fluorescence intensity in the middle of the gel (x = 500 μm). Dots represent measured values, solid line is based on diffusion calculations, with D = 1.33 × 10−6 cm2/s
The key strength of this approach is its simplicity, a setup that generates gel-filled microchannels in one step. The microfluidic nature of this assay allows the source and sink streams to be replenished continuously, maintaining stable gradient across microscopic 3D collagen gels without manual intervention.
4 Conclusions
We have developed a microfluidic Ladder Chamber capable of generating stable concentration gradients across 2D surfaces and 3D matrices. Neutrophil chemotaxis was successfully demonstrated in the Ladder Chamber. Compared to other assays, this chamber offers simplicity and improved stability of the gradients. In addition, the chamber provides a high-throughput approach for rapid characterization of chemotactic responses. The diffusion chamber also provides a straightforward method for generating concentration gradients in three-dimensional gels. This can be adapted to a variety of gels, allowing chemotaxis to be studied in different 3D environments. The simple nature of this approach, coupled with its versatility, will make it broadly applicable in cell migration research.
References
M. Bailly, L. Yan, G.M. Whitesides, J.S. Condeelis, J.E. Segall, Exp. Cell Res. 241, 285–299 (1998)
S. Boyden, J. Exp. Med. 115, 453–466 (1962)
X. Cao, M.S. Shoichet, Neuroscience 103, 831–840 (2001)
B.G. Chung, L.A. Flanagan, S.W. Rhee, P.H. Schwartz, A.P. Lee, E.S. Monuki, N.L. Jeon, Lab Chip 5, 401–406 (2005)
J.S. Condeelis, J.B. Wyckoff, M. Bailly, R. Pestell, D. Lawrence, J. Backer, J.E. Segall, Semin. Cancer Biol. 11, 119–128 (2001)
J. Crank, The Mathematics of Diffusion (Clarendon, Oxford, 1964)
E.L. Cussler, Diffusion: Mass Transfer in Fluid Systems (Cambridge University Press, New York, 1997)
S.K.W. Dertinger, D.T. Chiu, N.L. Jeon, G.M. Whitesides, Anal. Chem. 73, 1240–1246 (2001)
P.R. Fisher, R. Merkl, G. Gerisch, J. Cell Biol. 108, 973–984 (1989)
P. Friedl, P.B. Noble, K.S. Zanker, J. Immunol. Methods 165, 157–165 (1993)
P. Friedl, K.S. Zanker, E.B. Brocker, Microsc. Res. Tech. 43, 369–378 (1998)
G. Gerisch, H.U. Keller, J. Cell Sci. 52, 1–10 (1981)
S. Goswami, E. Sahai, J.B. Wyckoff, M. Cammer, D. Cox, F.J. Pixley, E.R. Stanley, J.E. Segall, J.S. Condeelis, Cancer Res. 65, 5278–5283 (2005)
J.L. Haddox, R.R. Pfister, C.I. Sommers, J. Immunol. Methods 141, 41–52 (1991)
J.L. Haddox, I.W. Knowles, C.I. Sommers, R.R. Pfister, J. Immunol. Methods 171, 1–14 (1994)
N.L. Jeon, S.K.W. Dertinger, D.T. Chiu, G.M. Whitesides, Langmuir 16, 8311–8316 (2000)
N.L. Jeon, H. Baskaran, S.K.W. Dertinger, G.M. Whitesides, L. Van De Water, M. Toner, Nat. Biotechnol. 20, 826–830 (2002)
S. Kanegasaki, Y. Nomura, N. Nitta, S. Akiyama, T. Tamatani, Y. Goshoh, T. Yoshida, T. Sato, Y. Kikuchi, J. Immunol. Methods 282, 1–11 (2003)
J. Kassis, D.A. Lauffenburger, T. Turner, A. Wells, Semin. Cancer Biol. 11, 105–119 (2001)
D.M. Knapp, E.F. Helou, R.T. Tranquillo, Exp. Cell Res. 247, 543–553 (1999)
D.A. Lauffenburger, S.H. Zigmond, J. Immunol. Methods 40, 45–60 (1981)
F. Lin, C.M. Nguyen, S.J. Wang, W. Saadi, S.P. Gross, N.L. Jeon, Biochem. Biophys. Res. Commun. 319, 576–581 (2004a)
F. Lin, W. Saadi, S.W. Rhee, S.-J. Wang, S. Mittal, N.L. Jeon, Lab Chip 4, 164–167 (2004b)
F. Lin, C.M. Nguyen, S.J. Wang, W. Saadi, S.P. Gross, N.L. Jeon, Ann. Biomed. Eng. 33, 475–482 (2005)
A.M. Lohof, M. Quillan, Y. Dan, M.M. Poo, J. Neurosci. 12, 1253–1261 (1992)
G. Maheshwari, A. Wells, L.G. Griffith, D.A. Lauffenburger, Biophys. J. 76, 2814–2823 (1999)
P.V. Moghe, R.D. Nelson, R.T. Tranquillo, J. Immunol. Methods 180, 193–211 (1995)
R.D. Nelson, P.G. Quie, R.L. Simmons, J. Immunol. 115, 1650–1656 (1975)
R.M. Nerem, R.W. Alexander, D.C. Chappell, R.M. Medford, S.E. Varner, W.R. Taylor, Am. J. Med. Sci. 316, 169–175 (1998)
W.J. Rosoff, J.S. Urbach, M.A. Esrick, R.G. McAllister, L.J. Richards, G.J. Goodhill, Nat. Neurosci. 7, 678–682 (2004)
W.J. Rosoff, R. McAllister, M.A. Esrick, G.J. Goodhill, J.S. Urbach, Biotechnol. Bioeng. 91, 754–759 (2005)
W. Saadi, S.-J. Wang, F. Lin, N.L. Jeon, Biomed. Microdevices 8, 109–118 (2006)
L. Soon, G. Mouneimne, J. Segall, J. Wyckoff, J. Condeelis, Cell Motil. Cytoskelet. 62, 27–34 (2005)
C. Tanford, Physical Chemistry of Macromolecules (Wiley, New York, 1961)
M.D. Tang, A.P. Golden, J. Tien, J. Am. Chem. Soc. 125, 12988–12989 (2003)
G.M. Walker, J. Sai, A. Richmond, M. Stremler, C.Y. Chung, J.P. Wikswo, Lab Chip 5, 611–618 (2005)
S.-J. Wang, W. Saadi, F. Lin, C.M.-C. Nguyen, N.L. Jeon, Exp. Cell Res. 300, 180–189 (2004)
A. Wells, Adv. Cancer Res. 78, 31–101 (2000)
A. Wells, J. Kassis, J. Solava, T. Turner, D.A. Lauffenburger, Acta Oncol. 41, 124–130 (2002)
G.M. Whitesides, E. Ostuni, S. Takayama, X. Jiang, D.E. Ingber, Annu. Rev. Biomed. Eng. 3, 335–373 (2001)
P.C. Wilkinson, J. Immunol. Methods 216, 139–153 (1998)
H. Wu, B. Huang, R.N. Zare, J. Am. Chem. Soc. 128, 4194–4195 (2006)
Z. Xiao, N. Zhang, D.B. Murphy, P.N. Devreotes, J. Cell Biol. 139, 365–374 (1997)
X. Yang, J. Corvalan, P. Wang, C. Roy, C. Davis, J. Leukoc. Biol. 66, 401–410 (1999)
J.Q. Zheng, M. Felder, J.A. Connor, M.M. Poo, Nature 368, 140–144 (1994)
D. Zicha, G. Dunn, A. Brown, J. Cell Sci. 99, 769–775 (1991)
S.H. Zigmond, J. Cell Biol. 75, 606–616 (1977)
Acknowledgment
We thank Professor Steven S. George and Justin Mih for reagents and valuable discussions. We also thank Dr. Kapil Krishan for assistance with diffusion calculations, and Dr. Mike Papac for equipment. This research was supported by the Department of Defense (Grants no. DAMD17-03-1-0515 and no. DAMD17-03-1-0673).
Author information
Authors and Affiliations
Corresponding author
Additional information
Equal contribution from Saadi and Rhee.
Supporting Information
Movie of neutrophil migration in a 0–50 ng/mL IL-8 gradient inside microgrooves will be available in supporting information.
Rights and permissions
About this article
Cite this article
Saadi, W., Rhee, S.W., Lin, F. et al. Generation of stable concentration gradients in 2D and 3D environments using a microfluidic ladder chamber. Biomed Microdevices 9, 627–635 (2007). https://doi.org/10.1007/s10544-007-9051-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10544-007-9051-9