
Abstract
Autophagy is a conserved mechanism that plays a housekeeping role by eliminating protein aggregates and damaged organelles. Recent studies have demonstrated that acute ethanol intoxication induces hepatic autophagy in mice. The effect of dietary zinc intake on hepatic autophagic flux during ethanol intoxication has not been evaluated using animal models. Herein, we investigated whether zinc deficiency and excess can affect autophagic flux in the liver in mice and in human hepatoma cells acutely exposed to ethanol. A mouse model of binge ethanol feeding was utilized to analyze the effect of low, adequate, and high zinc intake on hepatic autophagic flux during ethanol intoxication. Autophagic flux was inferred by analyzing LC3II/LC3I ratio, protein levels of p62/SQSTM1, Beclin1 and Atg7, and phosphorylation of 4EBP1. In addition, the degradation of the fusion protein LC3-GFP and the formation of autophagosomes and autolysosomes were evaluated in cells. Ethanol treatment stimulated autophagy in mice and cells. High zinc intake resulted in enhanced autophagy in mice exposed to ethanol. Conversely, zinc deficiency was consistently associated with impaired ethanol-induced autophagy in mice and cells. Zinc-deficient mice exhibited a high degree of ethanol-driven steatosis. Furthermore, zinc depletion increased apoptosis in cells exposed to ethanol. The results of this study suggest that adequate zinc intake is necessary for proper stimulation of autophagy by ethanol. Poor zinc status is commonly found among alcoholics and could likely contribute to faulty autophagy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is a conserved catabolic process used by eukaryotic cells for the degradation of damaged or superfluous proteins and organelles (Chen and Klionsky 2011). There are three forms of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy (Chen and Klionsky 2011). Among these three types of autophagy, macroautophagy (hereafter referred as autophagy) is the focus of this study. Recent studies with mice and cell models have shown that acute ethanol ingestion activates autophagy in liver (Ding et al. 2010; Ni et al. 2013; Lin et al. 2013). However, chronic ethanol intoxication seems to suppress hepatic autophagy (Thomes et al. 2015; Cho et al. 2014). The inhibition of autophagy has been shown to worsen ethanol driven steatosis and liver injury in mice (Ding et al. 2010; Ni et al. 2013; Lin et al. 2013). Conversely, pharmacological promotion of autophagy seems to alleviate ethanol-driven liver steatosis and liver injury (Lin et al. 2013). As a result, autophagy is regarded as a protective mechanism against the cytotoxic effects of ethanol and has emerged as an attractive target for the development of novel therapeutic agents for alcoholic liver disease (ALD). There is increasing evidence supporting the notion that zinc is critical for autophagy. It has been shown that zinc is essential for the induction of autophagy by tamoxifen in MCF-7 breast cancer cells and retinal cells, by hydrogen peroxide, hypoxia and re-oxygenation in astrocytes, and by ethanol in human hepatoma cells (Hwang et al. 2010; Hung et al. 2013; Cho et al. 2012; Pan et al. 2015; Lee et al. 2009; Liuzzi and Yoo 2013). Given the potential role of zinc in autophagy, studies addressing the effect of zinc intake on autophagy during acute or chronic elevated ethanol intake in animal models could help underscore the importance of adequate zinc intake in individuals with alcohol use disorder. Moreover, these studies could help develop novel therapeutic approaches to treat ALD. Binge drinking episodes are an example of acute exposure to elevated ethanol intake in humans. Binge drinking is common among Americans with an estimated 23.1% of Americans participating in binge drinking once a month (Substance Abuse and Mental Health Services Administration 2011). Although chronic alcohol abuse is commonly associated with illness caused by alcohol, acute alcohol abuse is also detrimental and can cause liver injury and steatosis (Massey and Arteel 2012). Notably, zinc deficiency is one of the most common nutritional deficiencies in alcoholics (McClain and Su 1983; Bode et al.1988). Poor zinc status in alcoholics is likely the consequence of the combined effect of inadequate zinc intake and altered metabolism of zinc (McClain and Su 1983;McClain et al. 1986; Dinsmore et al. 1985). It is significant that zinc supplementation has been observed to attenuate ethanol-induced liver injury in mice (Zhou et al. 2005; Kang et al. 2009).
In this study, we set out to examine the effect of zinc intake on hepatic autophagic flux mice exposed to acute ethanol intoxication. In addition, the role of zinc in autophagy during acute ethanol exposure was evaluated using an in vitro model of human hepatoma cells.
Methods
Animal study
A mouse model of binge ethanol feeding was employed (2). C57BL/6 male mice were purchased from Jackson Laboratories (Bar Harbor, ME). Male C57BL/6 mice have been previously used to evaluate the role of ethanol in the regulation of autophagy (Ding et al. 2010; Ni et al. 2013; Cho et al. 2014). Mice weighing 25–30 g were employed in the study. Mice were housed individually in plastic cages with raised wire floors. AIN-93 M semi-purified diets with casein replaced by 20% egg whites (Supplementary Table 1A) were utilized in this study. Egg whites were used as the source of protein instead of casein because of their lower zinc content (Stirn et al. 1935). Diets were purchased from LabDiet (St. Louis, MO). The zinc content of the diets was measured by inductively coupled plasma mass spectrometry (ICP-MS) using a Thermo 6500 dual view ICP instrument (Thermo Fisher Scientific Inc. Waltham, MA (Supplementary Table 1B). The dietary protocol utilized in this study to manipulate zinc status is similar to a previous study (Liuzzi et al. 2004). Conditions of zinc deficiency, adequacy and excess were generated in mice by feeding them diets containing three levels of elemental zinc for 28 days: low (1.0 mg/kg) (ZD), adequate (30 mg/kg) (AD), or supplemented (150 mg/kg) (SP). After this, mice were fasted for 4 h and administered either water or ethanol (4.5 g/kg body weight) by gavage (four gavages in 1 h) (Ding et al. 2010). There were six groups of mice: (1) low zinc (ZD), (2) low zinc and ethanol (ZDE), (3) adequate zinc (AD), (4) adequate zinc and ethanol (ADE), (5) excess zinc (SP), and (6) excess zinc and ethanol (SPE). A group of mice was injected intraperitoneally with either the autophagy inhibitor chloroquine (CQ) (dissolved in saline) (60 mg/kg body weight) or saline, 30 min prior to ethanol or water treatment (Ding et al. 2010). Mice were sacrificed by anesthesia overdose (Isoflurane (2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane)) at 24 h after ethanol treatment. This time point was selected because previous studies showed that ethanol stimulated autophagy in cells after 24 h (Ding et al. 2010; Liuzzi et al. 2004). The ethanol and CQ dosage proposed here were employed in a study in which the effect of acute ethanol exposure in autophagy was examined (Ding et al. 2010). The animal protocol was approved by the Institutional Laboratory Animal Care and Use Committee of Florida International University.
Analysis of autophagy
Autophagic flux was inferred by combining several markers: LC3II/LC3I ratio, protein levels of p62/SQSTM1, Beclin1 and Atg7, and phosphorylation of 4EBP1 (Klionsky et al. 2016). Increased LC3II/LC3I ratio and LC3-puncta are indicative of activation of autophagy. On the other hand, p62/SQSTM1 is known to be incorporated in autophagosomes and degraded in autolysosomes. Hence, a reduced p62/SQSTM1 protein level is considered as indicative of increased autophagy (Klionsky et al. 2016). Elevated expression of Beclin1 and Atg7 is indicative of autophagy activation (Klionsky et al. 2016). 4EBP1 is a target of mTOR, a serine/threonine kinase that is known to be a negative regulator of autophagy (Jung et al. 2010). Thus, low levels of phosphorylated 4EBP1 could reflect activation of autophagy. The formation of autophagosomes and autolysosomes (AV and AL) in cultured cells was analyzed. Increased formation of AV and AL suggests stimulation of autophagy. Lastly, the degradation of the fusion protein GFP-LC3, a substrate of autophagy (Klionsky et al. 2016), was evaluated in VL17A cells. The autophagy inhibitor CQ was employed to determine whether the results observed for LC3II/I ratio and AV/AL formation were caused by impaired lysosomal function rather than induction of autophagy (Klionsky et al. 2016).
Measurement of tissue triglyceride levels
Total triglyceride content in liver was assayed using the Triglyceride Colorimetric Assay Kit from Cayman Chemical (Ann Arbor, MI).
Histological analysis
Frozen sections of liver (10 micrometers) were stained with Oil Red O to examine steatosis. Steatosis in tissue sections was determined by the number and size of lipid droplets.
Alanine transaminase (ALT)
Liver injury was evaluated by measuring alanine transaminase (ALT) in serum. A colorimetric kit from Cayman Chemical was employed.
Zinc content in liver and diets
Zinc content in liver was analyzed by ICP-MS.
Cell culture and treatments
Experiments with human hepatoma VL17A cells were carried out to examine the effects of zinc deficiency, adequacy, and excess on autophagy. The VL17A cell line was generated from HepG2 cells (Donohue et al. 2006). VL17A cells were used because they have close to normal expression levels of two key alcohol-metabolizing enzymes: alcohol dehydrogenase (ADH) and cytochrome P450 (CYP2E1) (Donohue et al. 2006). VL-17A cells were kindly donated by Dr. Dahn Clemens (Department of Veterans Affairs, Nebraska Western Iowa). Cells were grown in a 5% CO2-enriched atmosphere in DMEM (GE Healthcare Life Sciences SH30285.01) medium containing 22 mM glucose supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 µg/ml streptomycin, and amphotericin B (Sigma, St. Louis, MO). Cells were seeded in 12 well plates (1.8 × 105 cells per well) and allowed to grow for 24 h before they were treated. To generate zinc deficiency and adequacy, cells were grown in either low zinc (0 µM) or adequate zinc (5 µM) cell culture medium for 48 h. To remove zinc from the medium, cell culture medium was prepared using fetal bovine serum (FBS) pretreated with the zinc chelating resin Chelex-100 (BioRad, Hercules, CA) (Ho and Ames 2002). The levels of zinc, iron and, copper in the medium were analyzed using ICP-MS (Perkin Elmer Sciex, Model ELAN DRC-II). Zinc was not detectable in medium prepared with fetal bovine serum pretreated with Chelex-100. Chelex-100 pretreatment did not affect iron and copper levels in the medium. Chelex-100 can also bind calcium and magnesium (Blankenship and Heitman 2005). DMEM medium (SH30285.01 (HycloneTM, Logan, UT)) contains 200.00 mg/L CaCl2 and 97.67 mg/L MgSO4. The average magnesium content in FBS is negligible as compared with the content in DMEM (Arigony et al. 2013). FBS is expected to contain 3–4 mM of calcium (Zoico et al. 2014). Thus, Chelex-100 treatment could lower calcium levels in the medium by 10%. However, this decrease in calcium levels in the medium should not affect the normal growth of cells. It has been shown that HepG2 cells can be cultured in either DMEM/F12 (50/50) or RPMI 1640 medium which have considerably lower calcium levels as compared to DMEM medium (Saliani et al. 2013; Joachimiak et al. 2007). ZnSO4 was added to zinc depleted medium to prepare medium with 5 µM zinc. Zinc deficient and zinc adequate cells were treated with or without ethanol for 24 h in medium without added l-glutamine. l-glutamine was removed from medium because it can affect basal and induced autophagy (Sakiyama et al. 2009). In some experiments, cells were exposed to the autophagy inhibitor CQ (30 µM) for 6 h. The effect of acute zinc treatment (24 h) on autophagy during ethanol exposure was also tested. Excess zinc medium was prepared by adding ZnSO4 to medium (prepared with FBS that was not pretreated with Chelex-100). The ethanol concentrations utilized in this study have been previously employed to evaluate the impact of ethanol exposure on hepatocytes (Ding et al. 2010; Ni et al. 2013; Liuzzi and Yoo 2013).
Generation of Atg7 knockout cell line
VL17-A cells were transfected with ATG7 CRISPR/Cas9 KO (h) and ATG7 HDR (h) plasmids from Santa Cruz Biotechnology. Stably transfected cells were selected by using puromycin (Santa Cruz Biotechnology, Dallas, TX). The use of the plasmids described above was approved by the Institutional Biosafety Committee of Florida International University.
Western Blot analysis
Liver samples and VL17-A cells were lysed in buffer (50 mM Tris and 2% Triton X-100) containing protease inhibitors (Halt protease inhibitor cocktail; Pierce, Rockford, IL). Cellular protein content was determined with a micro BCA protein assay kit (Pierce, Rockford, IL). Aliquots of homogenates were diluted with an equal volume of 5 × Laemmli sample buffer and resolved on 8-16% polyacrylamide gels (Genscript, Piscataway, NJ). Proteins were transferred onto nitrocellulose membranes using the eBlot® protein transfer system (Genscript, Piscataway, NJ). Anti-LC3B (MAP1LC3B) antibody was purchased from Novus Biologicals (cat. no. NB100-2220) (Littleton, CO), anti-p62/SQSTM1 (cat. no. 5114), anti-Beclin1 (BECN1) (cat. no. 3495), anti-Atg7 (cat. no. D12B11), anti-p4EBP1 (cat. no. 2855) and anti-4EBP1 (cat. no. 4923) were purchased from Cell Signaling Technology (Danvers, MA). GFP tag monoclonal antibody (GF28R) (cat. no. MA5-15256) was purchased from Thermofisher Scientific (Waltham, MA). Peroxidase conjugated goat anti-rabbit IgG and goat anti-mouse IgG (Santa Cruz Biotechnology) were used as secondary antibodies. Signal detection employed the SuperSignal West Pico chemiluminescent substrate (Thermofisher). The membranes were also probed with an anti-Actin antibody (cat. no. A2066) (Sigma, St. Louis, MO) to control for sample loading. The specificity of anti-Atg7, anti-LC3B, and anti-p62/SQSTM1 was confirmed using VL17A cells lacking Atg7. These cells have no expression of Atg7 and LC3II band. In addition, these cells have overexpression of p62/SQSTM1 due to lack of autophagy. Two bands were detected with anti-p62/SQSTM1 at approximately 40 and 60 kDa in VL17A cells lacking Atg7 and in liver samples. However, only the band at 40 kDa was consistently detected in cells expressing Atg7. The antibodies anti-Beclin1, anti-p4EBP1, and anti-4EBP1 exhibited the expected specific bands.
Assay of lactate dehydrogenase (LDH) activity
LDH release into the culture medium was assayed using the LDH cytotoxicity assay kit from Cayman Chemical.
Cyto-ID autophagosomes and autolysosomes (AV and AL) measurement and visualization
Cells were incubated in HBSS (Hank's Balanced Salt Solution) containing CYTO-ID (Enzo Life Sciences, Farmingdale, NY) and Hoechst (Immunochemistry Technologies) for 40 min. Thereafter, autophagosomes and autolysosomes (AV/AL) fluorescence was measured using a Biotek FLx800 plate reader (excitation 485 nm/emission 528 nm). Values were normalized to Hoechst fluorescence. Cyto-ID staining in cells was observed with a deconvolution microscopy system (Delta Vision imaging system, GE Healthcare Life-Sciences). The use of Cyto-ID to measure autophagy has been validated in previous reports (Guo et al. 2015; Chan et al. 2012).
Transient transfection of GFP-LC3 and RFP-LC3
VL17-A cells were transiently transfected with either the DNA construct pEX-GFP-hLC3WT or pEX-HcRed-hLC3WT. PEX-GFP-hLC3WT encoding green fluorescent protein (GFP) fused to human LC3B and pEX-HcRed-hLC3WT encoding HcRed fluorescent protein (RFP) fused to human LC3B. The plasmids containing these constructs were a gift from Tanida et al. (2008) (Addgene plasmids#24987 and 24991) (Cambridge, MA). The reagent Effectene (QIAGEN, Foster City, CA) was utilized for the transfections. The use of pEX-GFP-hLC3WT and pEX-HcRed-hLC3WT plasmids was approved by the Institutional Biosafety Committee of Florida International University.
mRNA quantitation by RT-PCR
Liver samples were homogenized in TRIzol (Life Technologies/Thermofisher, Grand Island, NY). Cells samples were lysed in TRIzol. Isolated RNA was treated with Turbo DNAse (Life Technologies/Thermofisher). Quantitative PCR (qPCR) was performed using the iScript™ one-step RT-PCR kit with SYBR ® Green (BioRad, Hercules, CA). The following set of primers were utilized for qPCR of human p62/SQSTM1: 5′- GCACCCCAATGTGATCTGC-3′ and 5′-CGCTACACAAGTCGTAGTCTGG-3′; mouse p62/SQSTM1: 5′-AGGATGGGGACTTGGTTGC-3′ and 5′-TCACAGATCACATTGGGGTGC-3′; human Actin 5′-CATGTAGGTTGCTATCCAGGC-3′ and 5′-CTCCTTAATGTCACGCACGAT-3′; and mouse Actin: 5′-TGTTACCAACTGGGACGACA-3′ and 5′-GGGGTGTTGAAGGTCTCAAA-3′. These primers were purchased from Life Technologies/Thermofisher. The sequences of primers were obtained from http://pga.mgh.harvard.edu/primerbank/. Predesigned primers (cat. no. QT02317819, Qiagen) were used to analyze MT2A (human Metallothionein 2A). Values were normalized to Actin.
Labile zinc and autophagosome/autolysosome (AV/AL) visualization
Cells were transfected with a plasmid encoding red fluorescence protein (RFP) and human MAP1LC3B gene. Transfected cells were treated with 20 µM zinc for 48 h and subsequently treated with 100 mM ethanol for 24 h. Thereafter, cells were incubated with incubated in HBSS containing the cell-permeable zinc-fluorophore NBD-TPEA (5μM) (Sigma-Aldrich). Labile zinc and AV/AL were observed using a deconvolution microscopy system (Delta Vision imaging system). Labile zinc was quantified using a Biotek FLx800 plate reader (excitation 485 nm/emission 528 nm). Values were normalized to Hoechst fluorescence.
Apoptosis measurement
Apoptosis was assayed using an Annexin V FITC apoptosis detection kit (BD Biosciences, San Jose, CA). Fluorescence was measured using a Biotek FLx800 plate reader (Winooski, VT). Values were normalized to Hoechst nuclear stain fluorescence (Immunochemistry Technologies, Bloomington, MN). Cells were observed with a deconvolution microscopy system (Delta Vision imaging system, GE Healthcare Life-Sciences, Pittsburgh, PA).
Statistical analysis
Data are presented as mean ± standard error (SE). Two-way ANOVA was used followed by Tukey’s post hoc test. Statistical significance was set at p < 0.05. Tests were performed using GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com.
Results
Food intake and weight gain
The dietary treatments did not affect food intake and body weight of mice (Supplementary Fig. 1, Supplementary Table 2).
Zinc liver content
The concentration of zinc in liver samples from zinc-deficient mice (ZD) was significantly lower than the concentration observed in samples from zinc-adequate (AD) (p < 0.05) and zinc-supplemented (SP) mice (p < 0.01) (Supplementary Fig. 2). However, the hepatic zinc concentration did not differ between AD and SP mice. Mice fed high zinc diet and ethanol (SPE) had significantly lower liver zinc content than SP mice.
Effect of zinc intake on hepatic autophagic flux in mice
Ethanol intake did not affect Atg7 expression (Fig. 1a, b). Low zinc intake was associated with decreased Atg7 expression in mice treated or not with ethanol (ZD and ZDE) as compared to the other groups, except for ADE mice (Fig. 1a, b). Two bands were detected by anti-p62/SQSTM1 antibody in liver samples. One band at 60 kDa and the other band at 40 kDa. The intensity of both bands was significantly decreased by ethanol treatment in zinc supplemented mice. However, only the 40 kDa band was significantly downregulated by ethanol in zinc adequate mice. Interestingly, ethanol treatment did not change p62/SQSTM1 protein expression in mice fed low zinc diets. There was a significant interaction between ethanol and zinc on p62/SQSTM1 protein expression. Noteworthy, p62/SQSTM1 mRNA levels were not affected by zinc status or ethanol treatment (Fig. 1b). Beclin1 expression was significantly increased in SPE mice as compared to zinc deficient and zinc adequate mice exposed or not to ethanol. LC3II/I ratio was increased by ethanol in zinc adequate (p < 0.05) and zinc supplemented mice (p < 0.0001). Nevertheless, this marker was not affected by ethanol in zinc deficient mice. Moreover, zinc status did not alter LC3II/I ratio under basal condition. Two-way ANOVA analysis of LC3II/I ratio revealed a very significant (p < 0.0001) interaction between zinc and ethanol. Lastly, the levels of phosphorylated 4EBP1 were significantly reduced in SPE mice as compared to ZD and ZDE mice.

Hepatic autophagic flux. a Markers of autophagic flux; b Western blot densitometry values of markers of autophagic flux and mRNA expression of p62/SQSTM1. Values are average of 4 to 6 mice per group ± SE. Means without a common letter are significantly different (p < 0.05). LC3II/I ratio: interaction zinc versus ethanol p < 0.0001. P62/SQSTM1(60 kDa): interaction zinc versus ethanol p < 0.0001
Hepatic steatosis, triglycerides, and liver injury
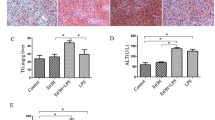
Ethanol binge feeding increased Oil Red O staining in ZDE, ADE and SPE mice. However, ZDE mice exhibited the highest degree of steatosis and triglyceride accumulation (Fig. 2a, b). Ethanol treatment significantly increased the concentration of hepatic triglycerides in ZDE (p < 0.001) and ADE (p < 0.01) mice but not in SPE mice. However, hepatic lipid accumulation did not vary between ADE and SPE mice. A significant interaction between zinc and ethanol was found for hepatic triglycerides (p < 0.0001). Interestingly, CQ treatment promoted ethanol-induced hepatic steatosis and lipid accumulation in ADE (p < 0.01) and SPE (p < 0.05) mice; however, it did not significantly affect hepatic triglyceride content in ZDE mice (Fig. 2a, c). Oil Red O staining showed that CQ decreased steatosis in ZDE mice. ALT levels in serum were increased by ethanol treatment in ZDE mice but not in ADE and SPE mice (Fig. 3a). CQ treatment did not increase ALT levels in mice (Fig. 3a, b).

Effect of zinc intake and autophagy on ethanol induced lipid accumulation in the liver. a Representative Oil Red O staining of the liver 20x; b and c concentration of hepatic triglycerides. Values are average of 4 mice per group ± SE. Means without a common letter are significantly different (p < 0.05). Interaction zinc versus ethanol p < 0.01. Interaction zinc versus CQ p < 0.05

Effect of zinc intake and autophagy on ethanol induced liver damage. a and b serum alanine aminotransferase (ALT). Values are average of 3 to 4 mice per group ± SE. * Significantly different (p < 0.05)
Effect of zinc depletion on autophagy and in human hepatoma cells
Beclin1 and Atg7 protein expression was not significantly affected by zinc and ethanol treatment (Fig. 4a, b). The levels of p62/SQSTM1 (40 kDa band) were significantly decreased by ethanol in zinc adequate cells (cultured in medium with 5 µM zinc) but not in zinc deficient cells (Fig. 4a, b). As expected, zinc depletion caused a drastic reduction in mRNA expression of the zinc regulated gene MT2A (Davis and Cousins 2000). However, p62/SQSTM1 mRNA expression was not affected by zinc or ethanol (Fig. 4c). The effect of zinc depletion on LC3II/LC3I ratio and p62/SQSTM1 expression in cells exposed to different levels of ethanol and was tested (Fig. 5a, b). LC3II/I ratio increased in zinc adequate cells exposed to 100 (p < 0.05) and 200 mM (p < 0.01) of ethanol. P62/SQSTM1 expression was reduced in zinc adequate cells treated with 50, 100 and 200 mM. There was a significant interaction (p < 0.05) between zinc and ethanol for LC3II/I ratio and p62/SQSTM1. CQ treatment resulted in increased LC3II/LC3I ratio in zinc-deficient and zinc-adequate cells exposed or not to ethanol (Fig. 6). To test whether the downregulation of p62/SQSTM1 in zinc adequate cells treated with ethanol is caused by stimulation of autophagy, we utilized VL17A cells lacking expression of the autophagy essential gene Atg7 (Atg7−/− cells). As expected, these cells exhibited elevated levels of p62/SQSTM1 as compared to wild type cells (Atg7+/+) (Fig. 7). Furthermore, the expression of p62/SQSTM1 in these cells was not influenced by zinc in medium or ethanol treatment (Fig. 7). Consistent with the notion that zinc is critical for autophagy, zinc depletion caused suppression of degradation of the fusion protein GFP-LC3 in cells exposed to ethanol (Fig. 8). The formation of AV/AL (as measured using Cyto-ID) was negatively affected by zinc depletion (Fig. 9a, b, e). Moreover, the addition of CQ caused accumulation of AV/AL in zinc-deficient and zinc-adequate cells treated with ethanol (Fig. 9c, d). Interestingly, the formation of AV/AL was found to be increased by zinc supplementation in a dose-dependent manner in cells treated with ethanol (Fig. 9f).

Effect zinc depletion on Atg7, Beclin1 and p4EBP expression in VL17-A cells. a Western blot images; b Western blot densitometry values of markers of autophagic flux; c mRNA expression of MT2A and p62/SQSTM1. Cells were incubated in zinc depleted medium (prepared with FBS treated with Chelex-100), supplemented with 0 or 5 µM zinc for 48 h and subsequently exposed to 0 or 100 mM ethanol for 24 h. Means without a common letter are significantly different (p < 0.05). * Significantly different (p < 0.05)

Effect of zinc deficiency and ethanol dosage on LC3II/I ratio and p62/SQSTM1 expression. a Western blot images; b Western blot densitometry values of LC3II/I ratio and p62/SQSTM1 expression. Cells were incubated in zinc depleted medium (prepared with FBS treated with Chelex-100), supplemented with 0 or 5 µM zinc for 48 h and subsequently exposed and exposed to various levels of ethanol. Values are average ± SE of three independent experiments. Means without a common letter are significantly different (p < 0.05). LC3II/I ratio interaction zinc versus ethanol p < 0.05. p62/SQSTM1 expression interaction zinc versus ethanol p < 0.05

Effect of chloroquine (CQ) on LC3II/I ratio in zinc deficient and zinc adequate cells treated or not with ethanol. Cells were treated as described in Fig. 4 legend. Some cells were treated with 30 µM of CQ for 6 h

Effect of zinc depletion on the protein expression of p62/SQSTM1 in autophagy deficient VL17A (cells lacking Atg7 expression). Cells were treated as described in Fig. 4 legend. Representative of three independent experiments

Degradation of GFP-LC3 fusion protein in VL17-A cells. Cells expressing GFP-LC3 fusion protein were treated as described in Fig. 4 legend. Representative of three independent experiments

Representative images of autophagosomes and autolysosomes (AV/AL). For a, b, c, d and e: cells were incubated in zinc depleted medium (prepared with FBS treated with Chelex-100), supplemented with 0 or 5 µM zinc for 48 h and subsequently exposed to 0 or 100 mM ethanol for 24 h. a Zinc depleted cells treated with 100 mM ethanol for 24 h; b cells treated with 5 µM and 100 mM ethanol for 24 h; c zinc depleted cells treated with 100 mM ethanol (24 h) and 30 µM CQ (6 h) and d cells treated with 5 µM of zinc, 100 mM ethanol, and 30 µM CQ (6 h). e and f Fluorimetric quantification of AV/AL utilizing CytoID. f Cells were incubated simultaneously with ethanol (100 mM) and different levels of zinc for 24 h (acute zinc treatment). Values are average ± SE of 3 to 4 independent experiments. Means without a common letter are significantly different (p < 0.05). Interaction zinc versus ethanol p < 0.05
Autophagosomes, autolysosomes (AV/AL) and free zinc in cells
The analysis of the intracellular localization of free zinc and AV/AL indicated a high degree of colocalization of labile zinc with these vesicles (Fig. 10a, b, c). Free zinc levels in cells were increased by zinc treatment. However, co-treatment with zinc and ethanol caused a further increase in free zinc levels in cells (Fig. 10d).

Autophagosomes and autolysosomes (AV/AL) and free zinc in VL17A cells. a, b and c: Colocalization of free zinc with autophagosomes and autolysosomes. Cells were treated with 20 µM of zinc for 48 h and subsequently treated with 100 mM of ethanol for 24 h. a Composite image showing colocalization of autophagosomes and autolysosomes with free zinc, b Free zinc (white arrows) and c autophagosomes and autolysosomes (white arrows). d Fluorimetric quantification of free zinc in VL17A cells. Cells were incubated simultaneously with ethanol and zinc for 24 h. Values are average ± SE of four independent experiments. Means without a common letter are significantly different (p < 0.05)
Effect of zinc depletion on apoptosis
Zinc-deficient cells exhibited increased ethanol induced apoptosis as compared to zinc-adequate cells (Fig. 11a, b). CQ treatment increased ethanol-induced apoptosis only in zinc-adequate cells (Fig. 11a, b). Of note, LDH leakage data in these cells indicated that the treatments did not cause noticeable increased cell death (Fig. 11c).

Zinc depletion enhances ethanol induced apoptosis and cell death through inhibition of autophagy. a Annexin V levels in VL17-A cells. b Representative images of Annexin V and Hoechst staining in VL17A cells. (I) Zinc depleted cells treated with 100 mM ethanol; (II) cells treated with 5 µM and 100 mM ethanol; (III) zinc depleted cells treated with 100 mM ethanol and CQ and (IV) cells treated with 5 µM of zinc, 100 mM ethanol, and CQ. c LDH leakage [percentage of “control group” defined as of cells treated with 5 µM zinc (no ethanol or CQ)]. Cells were treated as described in Fig. 4. Some cells were treated with 30 µM of CQ for 6 h. Values are average ± SE.* Significantly different (p < 0.05). Annexin V: interaction zinc versus chloroquine p < 0.0001
Discussion
In this study, we examined the effect of zinc deficiency, adequacy, and excess on autophagic flux under basal conditions and during acute ethanol exposure in the livers of mice and in human hepatoma cells. To our knowledge, this is the first study that demonstrates that zinc intake can affect autophagic flux in mice exposed to acute ethanol. The dietary treatments did not affect food intake or growth. Low zinc intake is known to cause anorexia in rats (Shay and Mangian 2000). However, mice did not seem to develop anorexia with mild to moderate zinc deficiency (Liuzzi et al. 2004). Consistent with previous studies (Ding et al. 2010; Ni et al. 2013; Lin et al. 2013), in our study, ethanol binge feeding stimulated hepatic autophagy. However, our results suggest that adequate zinc intake is needed for proper activation of autophagy by acute ethanol. Zinc deficient mice that were fed ethanol exhibited lower protein expression of the autophagy gene Atg7, decreased LC3II/I ratio as well as increased p62/SQSTM1 expression. These results indicate that low zinc intake had an inhibitory effect on the stimulation of autophagy by ethanol. Additionally, ZDE mice had increased steatosis and liver damage. On the other hand, our results suggest that zinc supplementation enhances ethanol induced autophagy. SPE mice showed increased LC3 II/I ratio and increased Beclin1 expression as compared to ADE mice. P62/SQSTM1 is an ubiquitin-binding autophagic adaptor protein that participates in the selective degradation of substrates by autophagy (Pankiv et al. 2007). Since p62/SQSTM1 is a substrate of autophagy, the levels of this protein inversely correlate with autophagic flux (Klionsky et al. 2016). Acute ethanol treatment decreased the protein levels of p62/SQSTM1 in ADE and SPE mice but not in ZDE mice. This suggests that zinc is likely a positive regulator of autophagic degradation of p62/SQSTM1. The fact that p62/SQSTM1 mRNA expression was not responsive to dietary zinc intake indicates that the observed effects on the protein expression of this gene are not likely to be due to the result of regulation of transcription or mRNA stability by zinc. Overall, the results from the studies with VL17A cells support the idea that zinc is critical for ethanol-induced autophagy. Zinc-deficient cells that were exposed to ethanol exhibited lower LC3II/I ratio and AV/AL formation and had increased levels of p62/SQSTM1 and phosphorylated 4EBP1 as compared to zinc-adequate cells treated with ethanol. The fact that CQ treatment increased LC3II/I ratio and AV/L in cells demonstrates that the observed differences in these markers were not due to impaired lysosomal function. Moreover, the fact that p62/SQSTM1 expression decreased concomitantly with an increase of LC3II/I ratio (in zinc adequate cells) also suggests that late autophagy was not inhibited. It is important to mention that although p62/SQSTM1 is mainly degraded by autophagy, this protein can be also degraded by the proteasome (Wong and Cuervo 2010). Based on the result that the expression of p62/SQSTM1 in autophagy deficient cells (Atg7−/− cells) was not affected by zinc or ethanol levels in the medium, it is unlikely that the observed results were caused by modulation of proteasomal degradation of p62/SQSTM1 by zinc. Consistent with the idea that zinc is a positive regulator of autophagy, the formation of AV/AL was negatively affected by zinc depletion and positively regulated by zinc in a dose-dependent manner. Furthermore, the inhibition of the degradation of the fusion protein GFP-LC3, a substrate of autophagy (Klionsky et al. 2016), by zinc depletion further supports the notion that zinc is critical for autophagy. The mechanisms by which ethanol induces autophagy are now beginning to be understood. Inhibition of mTOR and AKT, as well as activation of the transcription factors FOXO3A and TFEB, have been implicated in the activation of autophagy by acute ethanol (Ding et al. 2010; Ni et al. 2013; Thomes et al. 2015). Moreover, the oxidative stress generated by ethanol metabolization seems to be critical for the stimulation of autophagy by ethanol (Ding et al. 2010). The results of this study indicate that zinc depletion and zinc excess have a significant impact on autophagy during ethanol exposure. There was a consistent interaction between zinc and ethanol for LC3II/I ratio and p62/SQSTM1. In addition, ethanol caused a significant increase in free zinc levels in cells exposed to excess zinc. It is conceivable that the mobilization of zinc elicited by oxidative stress resulting from ethanol metabolization is an important component of the mechanism by which ethanol induces autophagy. Accumulating evidence from studies using “in vitro” models suggests that zinc modulates autophagy. It has been proposed that excess zinc could promote autophagy through phosphorylation of extracellular-signal-regulated kinases 1/2 (ERK1/2) and induction of the mRNA expression of autophagy genes (Hwang et al. 2010; Liuzzi and Yoo 2013; Wei et al. 2017). Moreover, zinc may play a role in AV/AL function. The colocalization of zinc with these vesicles found in this study and in a previous report (Lee et al. 2009) supports this hypothesis. Interestingly, a recent study showed that zinc depletion induced autophagy in yeast (Kawamata et al. 2017). Further studies are needed to determine what causes the differential response to zinc in yeast and mammalian cells. Reportedly, autophagy mitigates ethanol toxicity by promoting the degradation of lipid droplets (lipophagy), and mitochondria (mitophagy) (Ding et al. 2010). Zinc is known to play a protective role against ethanol toxicity (Zhou et al. 2005; Kang et al. 2009). Hence, it is an attractive hypothesis that some of the protective effects of zinc could be mediated through its role in autophagy. This idea is supported by the fact that the protective effects of adequate or high zinc intake against ethanol driven steatosis and liver injury were completely blocked by the autophagy inhibitor CQ. Lastly, the results of the analysis of early apoptosis in the presence/absence of CQ also support the idea that the modulation of autophagy by zinc may play an important role in the protective action of zinc against ethanol toxicity. Nevertheless, it is important to consider that CQ can also have unspecific effects. More studies are needed with other autophagy inhibitors to determine whether autophagy is involved in the protective effects of zinc. A recent study indicated that CQ is a zinc ionophore (Xue et al. 2014). Given the results of our study (colocalization of AV/AL with free zinc), it cannot be ruled out that the increased free zinc levels in cells caused by CQ, observed in that study, could be the result of AV/AL accumulation.
Autophagy is a stress response mechanism that is believed to confer protection against ethanol driven cytotoxicity. However, inadequate zinc intake can interfere with the regulation of autophagy by ethanol. Zinc deficiency is commonly found in alcoholics (McClain and Su 1983; Bode et al.1988); thus, poor zinc status is likely to be a contributing factor to faulty autophagy in alcoholics. Further studies are needed to establish the mechanisms by which zinc regulates autophagy. In addition, investigating the role of autophagy as a mediator of the protective effects of zinc could help develop novel therapeutic approaches to prevent alcoholic liver disease.
References
Arigony ALV, de Oliveira IM, Machado M et al (2013) The influence of micronutrients in cell culture: a reflection on viability and genomic stability. Biomed Res Int. https://doi.org/10.1155/2013/597282
Blankenship JR, Heitman J (2005) Calcineurin is required for Candida albicans to survive calcium stress in serum. Infect Immun 73:5767–5774
Bode JC, Hanisch P, Henning H, Koenig W, Richter FW, Bode C (1988) Hepatic zinc content in patients with various stages of alcoholic liver disease and in patients with chronic active and chronic persistent hepatitis. Hepatology 8:1605–1609
Chan LL-Y, Shen D, Wilkinson AR et al (2012) A novel image-based cytometry method for autophagy detection in living cells. Autophagy 8:1371–1382. https://doi.org/10.4161/auto.21028
Chen Y, Klionsky DJ (2011) The regulation of autophagy-unanswered questions. J Cell Sci 124(Pt 2):161–170
Cho KS, Yoon YH, Choi JA, Lee SJ, Koh JY (2012) Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Investig Ophthalmol Vis Sci 53:5344–5353. https://doi.org/10.1167/iovs.12-9827
Cho HI, Choi JW, Lee SM (2014) Impairment of autophagosome-lysosome fusion contributes to chronic ethanol-induced liver injury. Alcohol 48:717–725. https://doi.org/10.1016/j.alcohol.2014.08.006
Davis SR, Cousins RJ (2000) Metallothionein expression in animals: a physiological perspective on function. J Nutr 130:1085–1088
Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM (2010) Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 139:1740–1752
Dinsmore W, Callender ME, McMaster D, Todd SJ, Love AH (1985) Zinc absorption in alcoholics using zinc-65. Digestion 32:238–242
Donohue TM, Osna NA, Clemens DL (2006) Recombinant HepG2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol 38:92–101
Guo S, Liang Y, Murphy SF et al (2015) A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy 11:560–572. https://doi.org/10.1080/15548627.2015.1017181
Ho E, Ames BN (2002) Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFκB, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line. Proc Natl Acad Sci USA 99:16770–16775. https://doi.org/10.1073/pnas.222679399
Hung HH, Huang WP, Pan CY (2013) Dopamine- and zinc-induced autophagosome formation facilitates PC12 cell survival. Cell Biol Toxicol 29:415–429
Hwang JJ, Kim HN, Kim J, Cho DH, Kim MJ, Kim YS, Kim Y, Park SJ, Koh JY (2010) Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals 23:997–1013
Joachimiak R, Kaźnica A, Drewa T (2007) Influence of sodium butyrate on hepatocellular carcinoma (hepG2) and glioblastoma (C6) cell lines in vitro. Acta Pol Pharm 64:561–563
Jung CH, Ro S-H, Cao J, Otto NM, Kim D-H (2010) mTOR regulation of autophagy. FEBS Lett 584:1287–1295. https://doi.org/10.1016/j.febslet.2010.01.017
Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, Zhou Z (2009) Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology 50:1241–1250
Kawamata T et al (2017) Zinc starvation induces autophagy in yeast. J Biol Chem 292:8520–8530. https://doi.org/10.1074/jbc.M116.762948
Klionsky DJ et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12:1–222. https://doi.org/10.1080/15548627.2015.1100356
Lee SJ, Cho KS, Koh JY (2009) Oxidative injury triggers autophagy in astrocytes: the role of endogenous zinc. Glia 57:1351–1361. https://doi.org/10.1002/glia.20854
Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin XM (2013) Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 58:993–999
Liuzzi JP, Yoo C (2013) Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol Trace Elem Res 156:350–356. https://doi.org/10.1007/s12011-013-9816-3
Liuzzi JP, Bobo JA, Lichten LA, Samuelson DA, Cousins RJ (2004) Responsive transporter genes within the murine intestinal-pancreatic axis form a basis of zinc homeostasis. Proc Natl Acad Sci USA 101:14355–14360
Massey VL, Arteel GE (2012) Acute alcohol-induced liver injury. Front Physiol 3:193
McClain CJ, Su LC (1983) Zinc deficiency in the alcoholic: a review. Alcohol Clin Exp Res 7:5–10
McClain CJ, Antonow DR, Cohen DA, Shedlofsky SI (1986) Zinc metabolism in alcoholic liver disease. Alcohol Clin Exp Res 10:582–589
Ni H-M, Du K, You M, Ding W-X (2013) Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol 183:1815–1825
Pan R, Timmins GS, Liu W, Liu KJ (2015) Autophagy mediates astrocyte death during zinc-potentiated ischemia–reperfusion injury. Biol Trace Elem Res 166:89–95. https://doi.org/10.1007/s12011-015-0287-6
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24121–24145
Sakiyama T, Musch MW, Ropeleski MJ, Tsubouchi H, Chang EB (2009) Glutamine increases autophagy under basal and stressed conditions in intestinal epithelial cells. Gastroenterology 136:924–932. https://doi.org/10.1053/j.gastro.2008.12.002
Saliani N, Darabi M, Yousefi B et al (2013) PPARγ agonist-induced alterations in Δ6-desaturase and stearoyl-CoA desaturase 1: role of MEK/ERK1/2 pathway. World J Hepatol 5:220–225. https://doi.org/10.4254/wjh.v5.i4.220
Shay NF, Mangian HF (2000) Neurobiology of zinc-influenced eating behavior. J Nutr 130:1493S–1499S
Stirn FE, Elvehjem CA, Hart EB (1935) The indispensability of zinc in the nutrition of the rat. J Biol Chem 109:347–359
Substance Abuse and Mental Health Services Administration (2011) Results from the 2010 national survey on drug use and health: summary of national findings. Substance Abuse and Mental Health Services Administration, Rockville, MD. https://www.samhsa.gov/data/sites/default/files/NSDUHNationalFindingsResults2010-web/2k10ResultsRev/NSDUHresultsRev2010.pdf. Accessed 22 Dec 2017
Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K (2008) Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 4:131–134
Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr (2015) Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB. Alcohol Clin Exp Res 39:2354–2363. https://doi.org/10.1111/acer.12904
Wei CC, Luo Z, Song YF, Pan YX, Wu K, You WJ (2017) Identification of autophagy related genes LC3 and ATG4 from yellow catfish Pelteobagrus fulvidraco and their transcriptional responses to waterborne and diet borne zinc exposure. Chemosphere 175:228–238. https://doi.org/10.1016/j.chemosphere.2017.02.042
Wong E, Cuervo AM (2010) Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2:a006734. https://doi.org/10.1101/cshperspect.a006734
Xue J, Moyer A, Peng B, Wu J, Hannafon BN, Ding W-Q (2014) Chloroquine is a zinc ionophore. PLoS ONE 9:e109180. https://doi.org/10.1371/journal.pone.0109180
Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ (2005) Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol 166:1681–1690
Zoico E, Franceschetti G, Chirumbolo S, Rossi AP, Mazzali G, Rizzatti V, Budui S, Zamboni M (2014) Phenotypic shift of adipocytes by cholecalciferol and 1α,25 dihydroxycholecalciferol in relation to inflammatory status and calcium content. Endocrinology 155:4178–4188. https://doi.org/10.1210/en.2013-1969
Acknowledgements
This study was supported by NIH 1R03AA022451-01.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liuzzi, J.P., Narayanan, V., Doan, H. et al. Effect of zinc intake on hepatic autophagy during acute alcohol intoxication. Biometals 31, 217–232 (2018). https://doi.org/10.1007/s10534-018-0077-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-018-0077-7