
Abstract
Lactoferrin (LF) is an important antimicrobial and immune regulatory protein present in neutrophils and most exocrine secretions of mammals. The antimicrobial activity of LF has been related to the presence of an antimicrobial peptide sequence, called lactoferricin (LFcin), located in the N-terminal region of the protein. The antimicrobial activity of bovine LFcin is considerably stronger than the human version. In this work, chimera peptides combining segments of bovine and human LFcin were generated in order to study their antimicrobial activity and mechanism of action. In addition, the relevance of the conserved disulfide bridge and the resulting cyclic structure of both LFcins were analyzed by using “click chemistry” and sortase A-catalyzed cyclization of the peptides. The N-terminal region of bovine LFcin (residues 17–25 of bovine LF) proved to be very important for the antimicrobial activity of the chimera peptides against E. coli, when combined with the C-terminal region of human LFcin. Similarly the cyclic bovine LFcin analogs generated by “click chemistry” and sortase A preserved the antimicrobial activity of the original peptide, showing the significance of these two techniques in the design of cyclic antimicrobial peptides. The mechanism of action of bovine LFcin and its active derived peptides was strongly correlated with membrane leakage in E. coli and up to some extent with the ability to induce vesicle aggregation. This mechanism was also preserved under conditions of high ionic strength (150 mM NaCl) illustrating the importance of these peptides in a more physiologically relevant system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lactoferrin (LF) is an iron binding glycoprotein (80 kDa) that is present in most exocrine secretions of mammals (Alexander et al. 2012; Vogel 2012). LF is detected in high concentration in milk and particularly in human colostrum, where it is believed to play a protective role in nursing infants (Brock 1980; Sánchez et al. 1992). LF is also located in the secondary granules of neutrophils and constitutes an important element of the immune system, being involved in the pro- and anti-inflammatory response (Legrand et al. 2005; Latorre et al. 2012). In addition LF also exhibits antimicrobial activity against a wide range of pathogens, including bacteria, fungi and viruses (Valenti and Antonini 2005; Jenssen and Hancock 2009; Alexander et al. 2012). The ability of LF to chelate iron was initially considered as an important factor in its antimicrobial activity (Arnold et al. 1977, 1982), however LF directly interacts with the bacterial surface (Visca et al. 1990), and it induces the release of lipopolysaccharides (LPS) from the outer membrane. This perturbation of the bacterial surface affects the membrane permeability and renders the bacteria more susceptible to other antimicrobial agents (Jenssen and Hancock 2009).
The N-terminal lobe of LF exhibits a cluster of positively charged residues that play an important role in the ability of this protein to interact with negatively charged elements of the bacterial membrane (Baker and Baker 2009; Drago-Serrano et al. 2012). Furthermore, proteolytic digestion of LF by gastric-pepsin releases an antimicrobial peptide called lactoferricin (LFcin), which is located in the N-terminal region of the protein. In bovines, the lactoferricin peptide (LFcinB) is composed of residues 17–41 of bovine LF, while in humans the lactoferricin (LFcinH) is formed by residues 1–49 of human LF (Bellamy et al. 1992b; Hunter et al. 2005). The antimicrobial activity of both LFcins was considerably higher than the activity of the parent LF protein, indicating that these peptides are responsible for the antimicrobial activity of the entire protein. However, LFcinB was a stronger antibacterial and antifungal agent in vitro in comparison to LFcinH (Bellamy et al. 1992b; Tomita et al. 1994).
The mechanism of action of LFcinB is thought to involve the perturbation and permeabilization of the bacterial membrane. Evidence to support such a mechanism includes microscopy images of bacterial surface bleb formation, depolarization of the E. coli inner membrane (Ulvatne et al. 2001), potassium ion permeation in artificial bilayers (Umeyama et al. 2006) and permeation/collapse of E. coli and S. aureus membranes detected by atomic force microscopy (Liu et al. 2011). However an intracellular mechanism has also been proposed due to the low level of permeabilization observed in artificial membranes (Ulvatne et al. 2001), the inhibition of RNA synthesis (Ulvatne et al. 2004), and the internalization of LFcinB in bacterial cells (Haukland et al. 2001).
The lack of antimicrobial activity of LFcinH has been related to the length of the peptide and the differences in primary structure due to a relatively low sequence similarity (69 %) when compared to LFcinB (Gifford et al. 2005). Furthermore LFcinH and LFcinB exhibit different secondary structures in buffer and in hydrophobic environments. LFcinB is characterized by a β-sheet structure when interacting with lipid micelles (Hwang et al. 1998), while LFcinH acquires a partially α-helical conformation in a membrane mimetic solvent (Hunter et al. 2005; Gifford et al. 2005). Despite the primary and secondary structure differences, both LFcin peptides are characterized by the presence of a disulfide bridge, generating a distinctive cyclic loop. The importance of these disulfide bridges in the antimicrobial activity of the LFcin peptides is not completely clear. Several studies reported similar antimicrobial activities for native and linear peptides (Bellamy et al. 1992b; Liu et al. 2011), while another study showed a three times higher activity for the disulfide-bridged peptide in comparison to its linear counterpart (Vorland et al. 1998).
Cyclization of antimicrobial peptides (AMPs) has been used successfully in some cases to increase their bactericidal activity (Oren and Shai 2000; Dathe et al. 2004) or their serum stability (Nguyen et al. 2010). Several methodologies of cyclization have been developed, however its efficiencies can be highly variable (White and Yudin 2011). The application of the so-called “click chemistry” (a CuI-promoted alkyne/azide cycloaddition reaction) has emerged as a promising alternative for the design of peptide-based drugs and cyclization of antimicrobial peptides (Holland-Nell and Meldal 2011; Li et al. 2013). Another interesting method relies on the use of Staphylococcus aureus enzyme Sortase A. This enzyme recognizes the C-terminal LPETG sequence and in the presence of a N-terminal Gly residue induces the cyclization of the peptide, as previously shown for histatin-1 (Bolscher et al. 2011).
The present study focused on the antimicrobial activity and the mechanism of action of LFcinB and bovine–human chimeras. The chimera peptides were intended to improve the LFcinH activity as well as to contribute to our understanding of the mechanism of action of the LFcin-derived peptides. In addition, the antimicrobial activities of two new cyclic LFcinB analogs were analyzed and the effects on their mechanism of action were investigated. This study established that only LFcinB, its cyclic analogs and the chimera that included LFcinB17–25 exhibited a strong antimicrobial activity, which was linked to inner membrane permeabilization in E. coli. Changes in the ionic strength of the incubation media did not affect the peptide–membrane binding interactions, while the levels of membrane permeabilization were highly sensitive to the salt concentration, explaining the differences in antimicrobial activity among the peptides studied.
Materials and methods
Materials and bacterial strains
Escherichia coli ATCC 25922 was purchased from ATCC (Manassas, VA, USA) and E. coli ML35p was kindly provided by Dr. Robert Lehrer at UCLA David Geffen School of Medicine. Culture media Mueller–Hinton broth was obtained from Sigma-Aldrich (St. Louis, MO, USA) and tryptic soy broth was from Gibco Diagnostic (Madison, WI, USA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Solid-phase peptide synthesis
Linear peptides containing the segments required for cyclization (Table 1) were manufactured by solid-phase peptide synthesis using 9-fluorenylmethoxycarbonyl (Fmoc)-chemistry with a Syro II synthesizer (Biotage, Uppsala, Sweden) essentially as described previously (Bolscher et al. 2011). Peptide synthesis grade solvents were obtained from Actu-All Chemicals (Oss, The Netherlands), the preloaded NovaSyn TGA resins from NovaBiochem (Merck Schuchardt, Hohenbrunn, Germany) and the N-α-Fmoc-amino acids from Orpegen-Pharma (Heidelberg, Germany) and Iris Biotech (Marktredwitz, Germany). The azide and alkyne building blocks for click cyclization, i.e. Fmoc-L-azydolysine (Fmoc-Lys(N2)-OH) and Fmoc-L-bishomopropargylglycin (Fmoc-Bpg-OH), respectively, were obtained from Chiralix (Nijmegen, The Netherlands).
Peptide purification
Peptides were purified by preparative RP-HPLC on a Dionex Ultimate 3000 system (Thermo Scientific, Breda, The Netherlands) with a Grace Spring column 250 mm × 25 mm (Grace, Deerfield, IL, USA) containing Vydac C18 TP beads 10 μm (Hesperia, CA, USA). Elution was performed with a linear gradient from 15 to 45 % AcN containing 0.1 % TFA in 20 min at a flow rate of 20 ml/min. The absorbance of the column effluent was monitored at 214 nm, and peak fractions were pooled and lyophilized. Re-analysis by RP-HPLC on an analytic Vydac C18-column (218MS54) developed with a similar gradient at a flow rate of 1 ml/min revealed a purity of at least 95 %. The authenticity was confirmed by mass spectrometry with a Microflex LRF MALDI-TOF, equipped with a gridless reflectron (Bruker Daltonik GmbH, Bremen, Germany) as previously described (Bolscher et al. 2011).
Sortase-catalysed cyclization
The sortase-catalyzed cyclization was performed as described previously (Bolscher et al. 2011). Briefly, linear peptides were equipped with the sortase A cleavage site LPETGG at the C-terminus and a diglycine motif (GG) at the N-terminus, which served as a highly efficient acceptor allowing intra-molecular ligation (Table 1). The intra-molecular transpeptidation reactions were conducted with 0.5 mM of the linear peptide and 50 μM sortase A in sortase reaction buffer (50 mM Tris, pH 7.5, containing 150 mM NaCl and 10 mM CaCl2) at 37 °C, until cyclization was completed as indicated by a peak-shift in the RP-HPLC elution profile using an analytic Vydac C18-column (218MS54) developed with a 30 to 45 % AcN containing 0.1 % TFA in 20 min at a flow rate of 1 ml/min and a band-shift in SDS-PAGE, NU-PAGE 4–12 % gels (Life Technologies, Burlington, ON, Canada). The reaction mixture was subsequently separated by semi-preparative RP-HPLC on a Vydac C18 column (218MS510), eluted with a gradient from 30 to 45 % AcN containing 0.1 % TFA in 20 min at a flow rate of 4 ml/min. This resulted in separation of the cyclic peptide from the remnants of linear starting peptide, and from sortase A. Peak-fractions containing the cyclic peptide and sortase A, respectively, were pooled, lyophilized and stored as dry powder at −20 °C.
Click cyclization
The peptide cyclization using the click chemistry was based on a protein immobilization procedure described previously (Punna et al. 2005). Briefly, after synthesizing the linear LFcinB17–41 in which the Cys19 residue was replaced by Bpg-OH to introduce an alkyne and the Cys36 with a lysine residue in which the side group was terminated with an azide, the cyclization was performed on resin. To 4 eq. peptide in a 20 ml syringe, 8 eq. 2,6-lutidine, 8 eq. 2,2-bipyridine, 4 eq. CuBr and 8 eq. of sodium ascorbate (all peptide grade, Actu-All Chemicals, Oss, The Netherlands) were added and flushed for 1 min with N2. After incubation for 24 h at room temperature the mixture was flushed sequentially by DMF, H2O, MetOH, EDTA (100 mM), H2O and DMF and subsequently dried by flushing three times with isopropanol and DCM. Next the cyclized peptide was detached from the resin and purified as described (Bolscher et al. 2011).
Bovine–human peptide chimera synthesis
The bovine–human chimera lactoferricin peptides (Table 1) were purchased from 21st Century Biochemicals (Malboro, MA, USA). The peptides were synthesized by solid phase and the disulfide bridge between both cysteine residues was obtained through oxidation. Their purity (>95 %) and molecular weight were checked by HPLC and mass spectrometry, respectively.
E. coli antimicrobial activity
The minimal inhibitory concentration (MIC) of the lactoferricin-derived peptides against E. coli ATCC 25922 in MHB media was measured by the standard broth microdilution method previously described (Wiegand et al. 2008). Briefly E. coli cells were incubated at 5 × 105 cfu/ml in the presence of two-fold dilutions of MHB-dissolved peptides (0.25–128 μM) in a 96-well polypropylene plate and incubated for 18 h at 37 °C. To study the influence of salt concentration on the peptides MIC, a modified version of the MIC experiment was performed similarly as previously described (Krijgsveld et al. 2000; Park et al. 2000). E. coli cells were grown in MHB to 1 × 108 cfu/ml and diluted 100 times in NaP-TSB buffer [Na+-phosphate 10 mM pH 7.4 and TSB 1 % (v/v)] with or without addition of 150 mM NaCl. Cells at 5 × 105 cfu/ml were then incubated in the presence of two-fold dilutions of the peptides dissolved in the same media and incubated at 37 °C. After 3 h of incubation 100 μl of fresh MHB media was added and the cells were incubated for 18 h at 37 °C. The reported MICs correspond to the minimal peptide concentration where bacterial growth was not observed, for three independent experiments.
Large unilamellar vesicles (LUVs) preparation
LUVs composed of chicken egg derived phosphatidylglycerol (ePG) and phosphatidylethanolamine (ePE) at an equimolar ratio (1:1), or E. coli polar lipid extracts (PLE) were prepared by the extrusion method. Briefly, the necessary amount of the chloroform-dissolved lipids (Avanti Polar Lipids, Alabaster, AL, USA) were mixed in a glass vial and the organic solvent was initially evaporated under a stream of nitrogen. The remaining solvent was removed under vacuum overnight. The dry lipid films were resuspended in 500 μl of Tris-buffer (Tris 10 mM pH 7.4, EDTA 1 mM) supplemented with 30 or 150 mM NaCl by vigorous vortexing. The resuspended lipids were freeze-thawed seven times using liquid nitrogen followed by extrusion through two 0.1 μm polycarbonate filters (Nucleopore Filtration Products, Pleasanton, CA, USA) using a mini-extruder apparatus (Avanti Polar Lipids, Alabaster, AL, USA). The phospholipid concentration of LUVs was measured in triplicate by the Ames phosphate assay (Ames et al. 1966).
Tryptophan fluorescence and acrylamide quenching
The tryptophan fluorescence of the lactoferricin-derived peptides at 1 μM was measured in Tris-buffer supplemented with 30 or 150 mM NaCl in the absence or presence of 100 μM ePE:ePG LUVs, in a Varian Cary Eclipse fluorimeter (Varian Inc., Palo Alto, CA, USA) at 25 °C. The tryptophan residues were excited at 280 nm and the emission spectra registered between 300 and 500 nm, with a 10 nm slit width for excitation and emission. The change in the fluorescence emission maxima wavelength (blue shift) upon interaction of the peptides with LUVs at both salt concentrations was recorded.
Quenching of the tryptophan fluorescence was observed by eight sequential additions (15 μl/each) of the neutral quencher acrylamide (4 M) to 1 μM peptide, in the absence or presence of 100 μM LUVs, in Tris-buffer supplemented with 30 or 150 mM NaCl. The emission spectra were recorded after each acrylamide addition and the fluorescence intensity changes were analyzed through the Stern–Volmer plots and the quenching constant (Ksv) were calculated using the equation:
where Fo is the initial fluorescence intensity of the peptide and F is the fluorescence following addition of the quencher Q.
Aggregation of LUVs
The aggregation of LUVs, as induced by the lactoferricin-derived peptides, was registered spectrophometrically (Haney et al. 2012). PLE or ePE:ePG LUVs at 50 μM were incubated in the presence of peptides at different peptide to lipid molar ratios in a 96-well plate. The plates were incubated at 37 °C during 30 min with constant agitation (500 rpm). After incubation the absorbance at 400 nm was registered in a Thermo Scientific Genesys 10UV scanning spectrophotometer (Waltham, MA, USA).
E. coli inner membrane permeabilization
The permeabilization of the inner membrane in Gram-negative bacteria was studied as previously described (Epand et al. 2010). This method uses the E. coli strain ML35p, which constitutively expresses the intracellular enzyme β-galactosidase and lacks the lac permease. Hydrolysis of the membrane impermeable β-galactosidase substrate ortho-nitrophenyl-β-galactoside (ONPG) occurs upon permeabilization of the bacterial inner membrane and is registered by absorbance of the reaction product at 420 nm. E. coli ML35p was grown at 37 °C in LB media from a single colony until OD600 ~ 0.6. Next the cells were collected, washed and resuspended in NaP-TSB media or NaP-TSB supplemented with 150 mM NaCl. The cells were incubated at a final OD600 of 0.3 in the corresponding media, in the presence of ONPG (0.5 mM) and two-fold dilutions of the peptides (0–32 μM) in a 96-well plate. The absorbance in the wells was measured at 420 nm every 2 min, during 70 min in a Perkin Elmer Victor™ ×4 multilabel plate reader (Waltham, MA, USA) with shaking and temperature control at 37 °C.
Results
Antimicrobial activity
Bovine lactoferricin (LFcinB) exhibited a strong and salt dependent antimicrobial activity against E. coli (Table 2). This activity (MIC 32 μM) was significantly higher than the activity of the equivalent of human lactoferricin derivative (residues 18–42 of human LF) in MHB media (MIC > 128 μM). In order to test the influence of the ionic strength on the bactericidal activity of LFcin peptides, the MIC experiments were performed in NaP-TSB media (low ionic strength), or NaP-TSB supplemented with 150 mM NaCl (high ionic strength). A considerable increase in the antimicrobial activity of LFcinB was observed in the low ionic strength media (MIC 2–4 μM). In contrast, the addition of 150 mM NaCl reduced LFcinB’s activity (MIC 64 μM). Due to the lack of activity for LFcinH in MHB, the effects of ionic strength on this peptide were not determined.
In an attempt to enhance the antimicrobial activity of LFcinH several chimera peptides combining sections of the corresponding human and bovine lactoferricin peptides were prepared (Table 1). Only the peptide containing LFcinB17–25 exhibited antimicrobial activity (MIC 32–64 μM) similar to the LFcinB in MHB media (Table 2). All chimera peptides including B17–25 displayed a strong antimicrobial activity in the low ionic strength media. As expected in the presence of 150 mM NaCl, most chimera peptides lose their anti-E. coli activity. Only B17-25 retained a weak antimicrobial activity (MIC 128 μM) when incubated in the high ionic strength media.
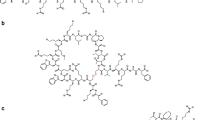
The use of “click chemistry”- and sortase-assisted cyclization of LFcinB did not considerably affect the antimicrobial potency of the peptides in comparison to the original disulfide-bridged LFcinB (Table 2). In the click peptide, the modification of the cysteine residues and the cyclization of the peptide occurs through a triazole link (Fig. 1), which did not affect its antimicrobial activity in MHB. In the high ionic strength media the MIC was two times lower. Contrary to the click peptide, LFcinB cyclized by sortase exhibited a slight increase in antimicrobial activity (MIC 16 μM) in MHB. This increase in activity was not only related to the five extra amino acid residues that are required for sortase recognition, as indicated by the results obtained for the linear version of the sortase-LFcinB peptide (MIC 32 μM). The ionic strength had a considerable effect on the antimicrobial activity of the sortase-cyclized peptide and its linear counterpart. Low NaCl concentration in the incubation media induced an increase in activity to 2–4 μM in MIC, while the presence of 150 mM NaCl reduced the antimicrobial activity to an MIC of 16–32 μM.

Amino acid sequences of LFcinB and its cyclic analogs. The residues X and Y represent the Bpg and Lys(N2), respectively, required for the click cyclization. The residues in purple correspond to mutated or additional residues required for sortase A recognition and cyclization
The presence of a loop motif in LFcinB, corresponding to residues 20–35 in LF (Fig. 1), warrants the question of whether the amino acids residues forming part of the closed loop will be sufficient to preserve the antimicrobial activity of the native LFcinB. The MIC experiments showed that the sortase-cyclic-loop (residues 20–35 and LPETGG) and its linear counterpart were unable to inhibit the growth of E. coli in MHB media and in NaP supplemented with 150 mM NaCl (Table 2). However, in low ionic strength media antimicrobial activity was observed, with MIC values of 32 and 16 μM for the linear and cyclic versions of these peptides, respectively.
Membrane interactions
It is expected that an increase in ionic strength in the incubation media will lead to a reduction in the electrostatic attractions between the negatively charged membranes of bacteria and the positively charged peptides due to screening effects (Nikaido 1998, 2003). We wanted to evaluate if the lack of antimicrobial activity at high salt concentrations was caused by a reduction of the peptide binding to membranes. Therefore the changes in the wavelength of the maximum tryptophan fluorescence for the LFcin-derived peptides (blue shift) in buffer and in the presence of LUVs were studied at low and high NaCl concentrations. Additionally the level of tryptophan exposure to the acrylamide quencher was evaluated under the same conditions. A mimetic system representing the negatively charged cytoplasmic membranes of Gram-negative bacteria was used. LUVs composed of ePE:ePG (1:1 molar ratio) were prepared in Tris-buffer supplemented with 30 or 150 mM NaCl, simulating both low and high ionic strength media, respectively. For the low ionic strength conditions, 30 mM NaCl had to be used in order to prevent destabilization of the ePG-containing vesicles.
In general all chimera and cyclic peptides exhibited a slightly larger blue shift when incubated in a low ionic strength media in comparison to the high ionic strength media (Fig. 2a). However this differences were not enough to explain the changes in antimicrobial activity. Interestingly the inactive sortase-cyclic-loop peptide exhibited the lowest blue shift in the presence of 150 mM NaCl.

Membrane interaction of LFcin peptides with ePE:ePG LUVs, followed by tryptophan fluorescence and acrylamide quenching. a Change in the fluorescence maxima wavelength (blue shift) upon peptide interaction with LUVs in Tris-buffer supplemented with 30 mM (black) or 150 mM NaCl (white). b Stern–Volmer constants (Ksv) for the peptides as determined by acrylamide quenching in different media: Tris-buffer supplemented with 30 mM (black) or 150 mM (white) NaCl, and LUVs in the same buffer supplemented with 30 mM (black dotted) and 150 mM (white dotted) NaCl. Results are average ± S.D. (n = 3)
The acrylamide quenching results were consistent with the outcome of the blue shift experiments (Fig. 2b). A high Ksv constant was calculated for all peptides in buffer, but no statistically significant differences in the Ksv were detected when comparing both ionic strength media. During incubation with ePE:ePG vesicles the Ksv was drastically reduced, indicating that the environment of the tryptophan residues shifted to a more hydrophobic surrounding upon interaction with the lipid bilayer. This reduction was also not dependent on the ionic strength of the buffer, and all chimera peptides exhibited similar Ksv values. For the cyclic peptides a slightly higher Ksv were observed in the presence of vesicles, with the higher value exhibited for the inactive cyclic peptide corresponding to the loop region of LFcinB.
Aggregation of LUVs
As established by the tryptophan fluorescence studies, all peptides interact with the ePE:ePG vesicles in low and high ionic strength media. However no direct correlation between the antimicrobial activity and membrane interaction was observed. Vesicle aggregation studies can be used as another indicator of membrane perturbation (Domingues et al. 2008). Hence the ability of the bovine–human chimera peptides and the cyclic LFcinB analogs to induce vesicles aggregation was studied by following the changes in absorbance at 400 nm (Fig. 3 and 4). When incubated in high ionic strength media only the native LFcinB and the chimera containing LFcinB17–23 exhibited ePE:ePG LUVs aggregation. However the LFcinB ability to induce aggregation was considerable higher. In contrast, the chimeras containing LFcinH18–24 and LFcinB17–25 only gave rise to limited aggregation at high peptide to lipid ratio, while the chimera containing LFcinB17–24 did not exhibit any aggregation (Fig. 3a). The reduction of the ionic strength only increased the vesicle aggregation of the peptides containing LFcinH18–24, B17–24 and B17–25 at high peptide concentration. In contrast, the activity of LFcinB and B17–23 was not affected (Fig. 3b). For the click and sortase-cyclic-LFcinB a strong ability to induce ePE:ePG vesicles aggregation was observed at high ionic strength (Fig. 3c). At lower ionic strength higher peptide concentrations were required to induce aggregation (Fig. 3d). In contrast, the cyclic peptide corresponding to the LFcinB loop region was not active (Fig. 3c and d).

ePE:ePG LUVs aggregation induced by LFcin peptides followed by the changes in absorbance at 400 nm. LUVs incubated in the presence of LFcinB and chimera peptides (a and b) or cyclic LFcinB analogs (c and d) in two different media, Tris-buffer supplemented with 30 mM (a and c) or 150 mM NaCl (b and d). LFcin peptides: LFcinB (filled diamond), B17–23 (filled square), B17–24 (asterisk), B17–25 (filled circle), H18–24 (filled triangle), Click-LFcinB (dotted line, filled diamond), SRT-cyclic-LFcinB (dotted line, filled triangle) and SRT-cyclic-Loop (dotted line, filled square). Results are average ± S.D. (n = 3)

PLE LUVs aggregation induced by LFcin peptides followed by the changes in absorbance at 400 nm. LUVs incubated in the presence of LFcinB and chimera peptides (a and b) or cyclic LFcinB analogs (c and d) in two different media, Tris-buffer supplemented with 30 mM (a and c) or 150 mM NaCl (b and d). LFcin peptides: LFcinB (filled diamond), B17–23 (filled square), B17–24 (asterisk), B17–25 (filled circle), H18–24 (filled triangle), Click-LFcinB (dotted line, filled diamond), SRT-cyclic-LFcinB (dotted line, filled triangle) and SRT-cyclic-Loop (dotted line, filled square). Results are average ± S.D. (n = 3)
LUVs composed of E. coli polar lipid extracts (PLE) were also prepared in order to more closely mimic the bacterial membrane. Although ePE:ePG vesicles constitute an acceptable initial bacterial membrane model, the composition of the PLE is slightly different with PE and PG accounting for 67 and 23.2 % respectively. Additionally PLE contains 9.8 % negatively charged cardiolipins. Using these vesicles, only LFcinB, click-LFcinB and sortase-cyclic-LFcinB were able to induce aggregation in the high ionic strength buffer (Fig. 4a and c). On the other hand, in the low ionic strength media all bovine–human chimera peptides exhibited an increase in the vesicle aggregation (Fig. 4b). Additionally, cyclic-click and sortase peptides required a higher peptide concentration in order to induce PLE vesicle aggregation. Again the cyclic loop peptide was not active in these experiments (Fig. 4c and d).
E. coli inner membrane permeabilization
The ability of LFcinB, chimeras and cyclic analogs to induce vesicle aggregation showed that membrane perturbation is related to the antimicrobial activity. However the mechanism of action might involve other processes distinct to vesicle aggregation such as membrane permeabilization. Here the permeabilization of bacterial membranes was studied by following the hydrolysis of the membrane impermeable β-galactosidase substrate ONPG (Epand et al. 2010). E. coli in the presence of LFcinB and incubated in a high ionic strength media (150 mM NaCl) exhibited a strong permeabilization of the inner membrane, as detected by a high rate of ONPG hydrolysis, even at peptide concentrations as low as 2 μM (Fig. 5, left). The low ionic strength conditions considerably increased the permeabilization of the inner membrane as observed by an increase in the rate of ONPG hydrolysis (Fig. 5, right). Most bovine–human chimeras caused rather low membrane permeabilization, considering that in comparison to LFcinB at least eight times higher peptide concentrations were required to observe ONPG hydrolysis in the high ionic strength media (Fig. 5, left). The peptide including LFcinB17–25 exhibited the strongest permeabilizing effect among the chimera peptides, with 8 μM being as active as 2 μM of LFcinB. In contrast when incubated in the low ionic strength media all chimera peptides exhibited similar and strong permeabilizing activities at concentrations as low as 1 μM. H18–24 exhibited a slightly higher rate of ONPG hydrolysis in these conditions (Fig. 5, right). One interesting difference between LFcinB and the chimera peptides was the time lag period before permeabilization/ONPG hydrolysis. LFcinB induces permeabilization after 2 min in low ionic strength conditions, while most chimeras required more than 10 min (Fig. 5, right).

Permeabilization of the E. coli inner membrane measured by ONPG hydrolysis, registering the absorbance at 420 nm. E. coli ML35p incubated in NaP-TSB buffer (right) or NaP-TSB supplemented with 150 mM NaCl (left) in the presence of LFcinB and bovine–human chimeras. Results are representative of three individual experiments
The click and sortase cyclic peptides exhibited a similar behavior to LFcinB, with fast and sizeable permeabilization of the E. coli inner membrane (Fig. 6). This permeabilizing effect was highly influenced by the ionic strength of the incubation media, being more pronounced at low ionic strength (Fig. 6, right). The sortase-cyclic-loop peptide in comparison only induced permeabilization in the low ionic strength media (Fig. 6, right), or at high concentrations when in high ionic strength media (Fig. 6, left).

Permeabilization of the E. coli inner membrane measured by ONPG hydrolysis, registering the absorbance at 420 nm. E. coli ML35p incubated in NaP-TSB buffer (right) or NaP-TSB supplemented with 150 mM NaCl (left) in the presence of cyclic LFcinB analogs and melittin. Results are representative of three individual experiments
In order to compare the behavior of the lactoferricin-derived peptides with a known membranolytic AMP, the permeabilization ability of melittin was also studied under the same conditions. This peptide induced the highest level of permeabilization, characterized by an extremely high rate of ONPG hydrolysis with a time delay of only 2 min when incubated with 150 mM NaCl. The reduction of the ionic strength further increased the rate of ONPG hydrolysis and reduced the lag period (Fig. 6, right).
Discussion
Antimicrobial activity of LFcin B and its mechanism of action
The mechanism of action of LFcinB is not yet completely understood. Several studies agree that the peptide displays a strong membrane activity in E. coli based on electron microscopy, inner membrane depolarization and atomic force microscopy, as well as K+ leakage measurements and studies of aggregation of vesicles (Ulvatne et al. 2001; Umeyama et al. 2006; Liu et al. 2011). However weak permeabilization seen with synthetic membranes and the lack of propidium iodide uptake in E. coli did not support such a membrane disruptive mechanism (Ulvatne et al. 2001). Consequently, a mechanism of action involving intracellular targets was also proposed, where LFcinB was found to be localized inside E. coli and S. aureus, and inhibited RNA synthesis at sub-lethal concentrations (Haukland et al. 2001; Ulvatne et al. 2004).
In this study the antimicrobial activity of LFcinB in MHB was between 3 and 16 times lower than the values previously reported (Bellamy et al. 1992a, b; Vorland et al. 1998). However the differences in bacterial strains and incubation media might account for this. The mechanism of action in our experiments was correlated with the ability of LFcinB to induce vesicle aggregation and to permeabilize the E. coli inner membrane (Figs. 3a, 4a and 5). The antimicrobial activity of LFcinB was markedly influenced by the ionic strength of the incubation media, however some activity was preserved under high ionic strength conditions, indicating that LFcinB can be a useful antimicrobial peptide under physiological conditions. Previous studies had established that LFcinB antimicrobial activity was modulated by the presence of monovalent (e.g. Na+ and K+) and divalent (e.g. Ca2+ and Mg2+) cations, however the specific mechanism being affected by the ionic strength was not studied (Bellamy et al. 1992a; Tomita et al. 1994). The current study shows that the difference in antimicrobial activity at low and high salt concentrations was related with changes in the membrane permeabilization induced by LFcinB (Fig. 5). Other parameters such as vesicle aggregation and peptide–membrane interactions did not correlate with the antimicrobial activity at different ionic strengths.
It was unanticipated that the increase in ionic strength did not affect the binding of the peptide to the artificial membranes (Fig. 2). Our fluorescence experiments indicated similar changes in the tryptophan fluorescence maxima wavelength (blue shift) upon interaction with ePE:ePG vesicles in the presence of 30 or 150 mM NaCl (Fig. 2a). In addition the quenching of the tryptophan fluorescence by acrylamide established that the solvent exposure of the LFcinB’s tryptophans was highly reduced when in the presence of LUVs in both ionic strength media (Fig. 2b). Together, these results indicated that peptide–membrane interactions were taking place and that the tryptophan residues were embedded in the bilayer, independently of the salt concentration of the medium. Therefore, these results suggest that the hydrophobic interactions could be driving the interaction between the peptides and membranes when in the presence of high salt concentrations.
A similar effect in vesicle aggregation was observed for LFcinB in both ionic strength conditions (Figs. 3 and 4). Considerable aggregation was observed after the incubation of ePE:ePG or PLE vesicles with LFcinB in both high and low ionic strength media. The only difference detected involved the minimal peptide concentration required to trigger aggregation. This could be related to an increase in the zeta-potential of the vesicle when incubated in the low ionic strength media, increasing the electrostatic repulsion among the vesicles (Crommelin 1984). Under these conditions more vesicle-bound peptide would be required to shield the negative charges of the vesicles, reducing their electrostatic repulsion and allowing aggregation.
Bovine–human LFcin chimeras
The construction of the chimera peptides based on the N- and C-terminal regions of bovine and human LFcins (Table 1), revealed that the N-terminal region of LFcinB was the most important peptide segment to preserve the antimicrobial activity of the native LFcinB. This N-terminal region needed to encompass residues 17–25 of LFcinB in order to exhibit maximal antimicrobial activity (Table 2). This result is in agreement with several studies showing strong antimicrobial activity of the LFcinB derived peptides containing the residues 17–30, 17–31 and the short hexapeptide RRWQWR after C-terminal amidation (Tomita et al. 1994; Strøm et al. 2002; Bolscher et al. 2009).
Differences in the charge of the peptides could not account for the differences in their antimicrobial activity or in the sensitivity to ionic strength. Similar to the peptide containing LFcinB17–25, the peptide containing LFcinB17–23 had the same net positive charge (+8) but exhibited poor antimicrobial activity (Table 2). The high antimicrobial activity of the peptide containing LFcinB17–25, similarly to the parent LFcinB peptide, was correlated with the ability of the peptide to induce membrane permeabilization. However the level of membrane permeabilization was considerably smaller in comparison to the original LFcinB peptide. Also the onset of ONPG hydrolysis was delayed when compared with the permeabilization profile of LFcinB. This time lag can be related with a slower transition of B17–25 from the bacterial outer surface to the inner membrane. It is interesting to note that all other chimera peptides (H18–24, B17–23 and B17–24) induced smaller degrees of inner membrane permeabilization, explaining the lack of antimicrobial activity at high ionic strength. In contrast, the ability to induce vesicle aggregation was only correlated with the antimicrobial activity when the vesicle system was composed of PLE lipids in the presence of 150 mM NaCl (Fig. 4). These results stress the importance of membrane composition when studying the peptide–membrane interactions.
Cyclic peptides
Replacing the disulfide bridge by a triazole during the “click” cyclization of LFcinB did not affect its antimicrobial activity. The click version of the peptide inhibited the growth of E. coli in a similar manner as the disulfide-bridged LFcinB (Table 2). Furthermore the mechanism of action was also preserved as indicated by the vesicle aggregation and E. coli permeabilization (Figs. 3, 4 and 6). These results support the use of triazole as a stable substitute for the disulfide bridge as was previously described for tachyplesyn I (Holland-Nell and Meldal 2011).
The use of sortase A provided an alternative route to generate cyclic LFcinB peptides without affecting the antimicrobial activity. In fact, in the case of sortase-cyclic-LFcinB the double loop structure generated by the cyclization of native LFcinB created a peptide with a slightly better antimicrobial activity (Table 2). The antimicrobial mechanism of this peptide also indicated a strong ability to induce vesicle aggregation and permeabilization of the E. coli inner membrane (Figs. 3, 4 and 6). On the other hand, the N- and C-terminal amino acid residues in LFcinB proved to be very important for its activity. The replacement of the sequences VRRAF and FK by the sortase recognition sequences LPET and GG completely abolished the antimicrobial activity of the sortase-cyclic-loop peptide (Table 2). In this case the lack of antimicrobial activity was expected due to the loss of four positive charges resulting in inability to interact with ePE:ePG vesicles, as showed by smaller blue shifts and larger Ksv when in the presence of vesicles (Fig. 2). Additionally, the poor vesicle aggregation capabilities and a lower ability to permeabilize the E. coli inner membrane also explained the lack of antimicrobial activity (Figs. 3, 4 and 6).
Abbreviations
- AMPs:
-
Antimicrobial peptides
- Bpg:
-
Bishomopropargylglycin
- ePG:
-
Egg derived phosphatidylglycerol
- ePE:
-
Egg derived phosphatidylethanolamine
- LF:
-
Lactoferrin
- LFcin:
-
Lactoferricin
- LPS:
-
Lipopolysaccharides
- LUVs:
-
Large unilamellar vesicles
- Lys(N2):
-
Azidolysine
- MIC:
-
Minimal inhibitory concentration
- MHB:
-
Mueller–Hinton broth
- ONPG:
-
Ortho-nitrophenyl-β-galactoside
- PLE:
-
Polar lipid extracts
- TSB:
-
Tryptic soy broth
References
Alexander DB, Iigo M, Yamauchi K, Suzui M (2012) Lactoferrin: an alternative view of its role in human biological fluids. Biochem Cell Biol 90:279–306. doi:10.1139/O2012-013
Ames BN, Neufeld EF, Ginsberg V (1966) Assay of inorganic phosphate, total phosphate and phosphatases. Methods Enzymol 8:115–118. doi:10.1016/0076-6879(66)08014-5
Arnold RR, Cole MF, McGhee JR (1977) A bactericidal effect for human lactoferrin. Science 197:263–265
Arnold RR, Russell JE, Champion WJ et al (1982) Bactericidal activity of human lactoferrin: differentiation from the stasis of iron deprivation. Infect Immun 35:792–799
Baker EN, Baker HM (2009) A structural framework for understanding the multifunctional character of lactoferrin. Biochimie 91:3–10. doi:10.1016/j.biochi.2008.05.006
Bellamy W, Takase M, Wakabayashi H et al (1992a) Antibacterial spectrum of lactoferricin B, a potent bactericidal peptide derived from the N-terminal region of bovine lactoferrin. J Appl Bacteriol 73:472–479
Bellamy W, Takase M, Yamauchi K et al (1992b) Identification of the bactericidal domain of lactoferrin. Biochim Biophys Acta 1121:130–136
Bolscher JGM, Adão R, Nazmi K et al (2009) Bactericidal activity of LFchimera is stronger and less sensitive to ionic strength than its constituent lactoferricin and lactoferrampin peptides. Biochimie 91:123–132. doi:10.1016/j.biochi.2008.05.019
Bolscher JGM, Oudhoff MJ, Nazmi K et al (2011) Sortase A as a tool for high-yield histatin cyclization. FASEB J 25:2650–2658. doi:10.1096/fj.11-182212
Brock JH (1980) Lactoferrin in human milk: its role in iron absorption and protection against enteric infection in the newborn infant. Arch Dis Child 55:417–421
Crommelin DJA (1984) Influence of lipid composition and ionic strength on the stability of liposomes. J Pharm Sci 73:1559–1563
Dathe M, Nikolenko H, Klose J, Bienert M (2004) Cyclization increases the antimicrobial activity and selectivity of arginine- and tryptophan-containing hexapeptides. Biochemistry 43:9140–9150. doi:10.1021/bi035948v
Domingues MM, Castanho MARB, Santos NC (2008) What can light scattering spectroscopy do for membrane-active peptide studies? J Pept Sci 14:394–400. doi:10.1002/psc
Drago-Serrano ME, de la Garza-Amaya M, Luna JS, Campos-Rodríguez R (2012) Lactoferrin–lipopolysaccharide (LPS) binding as key to antibacterial and antiendotoxic effects. Int Immunopharmacol 12:1–9. doi:10.1016/j.intimp.2011.11.002
Epand RF, Pollard JE, Wright JO et al (2010) Depolarization, bacterial membrane composition, and the antimicrobial action of ceragenins. Antimicrob Agents Chemother 54:3708–3713. doi:10.1128/AAC.00380-10
Gifford JL, Hunter HN, Vogel HJ (2005) Lactoferricin: a lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol Life Sci 62:2588–2598. doi:10.1007/s00018-005-5373-z
Haney EF, Nazmi K, Bolscher JGM, Vogel HJ (2012) Structural and biophysical characterization of an antimicrobial peptide chimera comprised of lactoferricin and lactoferrampin. Biochim Biophys Acta 1818:762–775. doi:10.1016/j.bbamem.2011.11.023
Haukland HH, Ulvatne H, Sandvik K, Vorland LH (2001) The antimicrobial peptides lactoferricin B and magainin 2 cross over the bacterial cytoplasmic membrane and reside in the cytoplasm. FEBS Lett 508:389–393
Holland-Nell K, Meldal M (2011) Maintaining biological activity by using triazoles as disulfide bond mimetics. Angew Chemie 50:5204–5206. doi:10.1002/anie.201005846
Hunter HN, Demcoe AR, Jenssen H et al (2005) Human lactoferricin is partially folded in aqueous solution and is better stabilized in a membrane mimetic solvent. Antimicrob Agents Chemother 49:3387–3395. doi:10.1128/AAC.49.8.3387
Hwang PM, Zhou N, Shan X et al (1998) Three-dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry 37:4288–4298. doi:10.1021/bi972323m
Jenssen H, Hancock REW (2009) Antimicrobial properties of lactoferrin. Biochimie 91:19–29. doi:10.1016/j.biochi.2008.05.015
Krijgsveld J, Zaat SAJ, Van Veelen PA et al (2000) Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem 275:20374–20381
Latorre D, Berlutti F, Valenti P et al (2012) LF immunomodulatory strategies: mastering. Biochem Cell Biol 90:269–278. doi:10.1139/O11-059
Legrand D, Elass E, Carpentier M, Mazurier J (2005) Lactoferrin: a modulator of immune and inflammatory responses. Cell Mol Life Sci 62:2549–2559. doi:10.1007/s00018-005-5370-2
Li H, Aneja R, Chaiken I (2013) Click chemistry in peptide-based drug design. Molecules 18:9797–9817. doi:10.3390/molecules18089797
Liu Y, Han F, Xie Y, Wang Y (2011) Comparative antimicrobial activity and mechanism of action of bovine lactoferricin-derived synthetic peptides. Biometals 24:1069–1078. doi:10.1007/s10534-011-9465-y
Nguyen LT, Chau JK, Perry NA et al (2010) Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 5:11–18. doi:10.1371/journal.pone.0012684
Nikaido H (1998) The role of outer membrane and efflux pumps in the resistance of gram-negative bacteria. Can we improve drug access? Drug Resist Updat 1:93–98. doi:10.1016/S1368-7646(98)80023-X
Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. doi:10.1128/MMBR.67.4.593
Oren Z, Shai Y (2000) Cyclization of a cytolytic amphipathic alpha-helical peptide and its diastereomer: effect on structure, interaction with model membranes, and biological function. Biochemistry 39:6103–6114. doi:10.1021/bi992408i
Park CB, Yi KS, Matsuzaki K et al (2000) Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: the proline hinge is responsible for the cell-penetrating ability of buforin II. Proc Natl Acad Sci USA 97:8245–8250. doi:10.1073/pnas.150518097
Punna S, Kaltgrad E, Finn MG (2005) “Clickable” agarose for affinity chromatography. Bioconjug Chem 16:1536–1541. doi:10.1021/bc0501496
Sánchez L, Calvo M, Brock JH (1992) Biological role of lactoferrin. Arch Dis Child 67:657–661
Strøm MB, Haug BE, Rekdal Ø et al (2002) Important structural features of 15-residue lactoferricin derivatives and methods for improvement of antimicrobial activity. Biochem Cell Biol 80:65–74. doi:10.1139/O01-236
Tomita M, Takase M, Bellamy W, Shimamura S (1994) A review: the active peptide of lactoferrin. Acta Paediatr Jpn 36:585–591
Ulvatne H, Haukland HH, Olsvik O, Vorland LH (2001) Lactoferricin B causes depolarization of the cytoplasmic membrane of Escherichia coli ATCC 25922 and fusion of negatively charged liposomes. FEBS Lett 492:62–65
Ulvatne H, Samuelsen Ø, Haukland HH et al (2004) Lactoferricin B inhibits bacterial macromolecular synthesis in Escherichia coli and Bacillus subtilis. FEMS Microbiol Lett 237:377–384. doi:10.1016/j.femsle.2004.07.001
Umeyama M, Kira A, Nishimura K, Naito A (2006) Interactions of bovine lactoferricin with acidic phospholipid bilayers and its antimicrobial activity as studied by solid-state NMR. Biochim Biophys Acta 1758:1523–1528. doi:10.1016/j.bbamem.2006.06.014
Valenti P, Antonini G (2005) Lactoferrin: an important host defence against microbial and viral attack. Cell Mol Life Sci 62:2576–2587. doi:10.1007/s00018-005-5372-0
Visca P, Dalmastri C, Verzili D et al (1990) Interaction of lactoferrin with Escherichia coli cells and correlation with antibacterial activity. Med Microbiol Immunol 179:323–333
Vogel HJ (2012) Lactoferrin, a bird’s eye view. Biochem Cell Biol 90:233–244. doi:10.1139/O2012-016
Vorland LH, Ulvatne H, Andersen J et al (1998) Lactoferricin of bovine origin is more active than lactoferricins of human, murine and caprine origin. Scand J Infect Dis 30:513–517
White CJ, Yudin AK (2011) Contemporary strategies for peptide macrocyclization. Nat Chem 3:509–524. doi:10.1038/nchem.1062
Wiegand I, Hilpert K, Hancock REW (2008) Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175. doi:10.1038/nprot.2007.521
Acknowledgments
The authors would like to thank Dr. Douglas Storey from the University of Calgary for the use of his laboratory facilities for the study of E. coli membrane permeabilization. This work has been funded by an operating grant from the “Novel alternatives to antibiotics” program of the Canadian Institutes of Health Research to Dr. Hans J. Vogel. Mauricio Arias and Dr. Hans J. Vogel were the holders of a Studentship and a Scientist award, respectively, of Alberta Innovates—Health Solutions (AIHS). Kamran Nazmi and Dr. Jan G.M. Bolscher were supported by a grant from the University of Amsterdam for research into the focal point Oral infections and Inflammation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Arias, M., McDonald, L.J., Haney, E.F. et al. Bovine and human lactoferricin peptides: chimeras and new cyclic analogs. Biometals 27, 935–948 (2014). https://doi.org/10.1007/s10534-014-9753-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-014-9753-4