
Abstract
Cadmium (Cd) is one of the most important environmental pollutants that cause a number of adverse health effects in humans and animals. Recent studies have shown that Cd-induced oxidative damage within the vascular tissues results in vascular dysfunction. The current study was aimed to investigate whether ascorbic acid could protect against Cd-induced vascular dysfunction in mice. Male ICR mice were received CdCl2 (100 mg/l) via drinking water for 8 weeks alone or received ascorbic acid supplementation at doses of 50 and 100 mg/kg/day for every other day. Results showed that Cd administration increased arterial blood pressure and blunted the vascular responses to vasoactive agents. These alterations were related to increased superoxide production in thoracic aorta, increased urinary nitrate/nitrite, increased plasma protein carbonyl, elevated malondialdehyde (MDA) concentrations in plasma and tissues, decreased blood glutathione (GSH), and increased Cd contents in blood and tissues. Ascorbic acid dose-dependently normalized the blood pressure, improved vascular reactivities to acetylcholine (ACh), phenylephrine (Phe) and sodium nitroprusside (SNP). These improvements were associated with significant suppression of oxidant formation, prevention of GSH depletion, and partial reduction of Cd contents in blood and tissues. The findings in this study provide the first evidence in pharmacological effects of ascorbic acid on alleviation of oxidative damage and improvement of vascular function in a mouse model of Cd-induced hypertension and vascular dysfunction. Moreover, our study suggests that dietary supplementation of ascorbic acid may provide beneficial effects by reversing the oxidative stress and vascular dysfunction in Cd-induced toxicity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd) is widely used in industry and also found as an important side contaminant in agricultural products. Cd contamination in soil and water has raised concerns because this metal is bio-accumulated in the upper levels of the food chain (WHO 1993; Satarug et al. 2010). In addition to occupational exposure, the two main sources of non-occupational exposure to Cd are cigarette smoking and diet (IARC 1993; Satarug and Moore 2004). Evidently, Cd content in the human body is likely to increase in the future and might lead to a higher incidence of Cd-related diseases including cardiovascular disease, hypertension, osteoporosis, nephrotoxicity, hepatotoxicity, diabetes, and cancers of many organs (Fowler 2009; Jarup and Akesson 2009; Satarug et al. 2010). Interestingly, vascular endothelium has been suggested to be a critical target of Cd toxicity which leads to many cardiovascular complications (Nakagawa and Nishijo 1996; Prozialeck et al. 2006, 2008; Messner and Bernhard 2010) Therein, Cd has been reported to be a possible risk factor of hypertension in experimental studies, such as renal tubular damage and dysfunction caused by environmental Cd exposure (Satarug et al. 2005). The hypertensive effect of Cd is resulted from complex actions on both the vascular endothelium and vascular smooth muscle cells (VSMCs). Cd, at relatively low, sublethal concentrations, increases oxidative stress and attacks endothelium and VSMCs at a variety of molecular levels, thereby leads to vascular damage and dysfunction (Prozialeck et al. 2008; Messner and Bernhard 2010).
Many studies connect Cd-intake with oxidative stress because several lines of evidences indicate that reactive oxygen species (ROS) and reactive nitrogen species (RNS) formed in the presence of Cd could be responsible for its toxic effects in many cells and tissues (Wang et al. 2004). ROS-induced by Cd directly oxidize lipids, proteins, and nucleic acids which lead to damage of the basic cell structures and result in cellular dysfunction and cell death (Bertin and Averbeck 2006; Kitamura and Hiramatsu 2010; Thevenod 2010). It has been demonstrated that Cd-induced oxidative stress is attenuated by antioxidants (Yokouchi et al. 2008). Therefore, supplementation of antioxidant should be one of the important components of an effective treatment of Cd poisoning.
Ascorbic acid, or vitamin C, is widely distributed in natural products, mostly rich in fresh fruits and leafy vegetables (Haytowitz 1995). Generally, ascorbic acid is regarded as the most important water-soluble antioxidant in plants, animals and humans (Mandl et al. 2009). Epidemiological studies propose the possible beneficial effects of vitamin C supplementation for the prevention of cancer and cardiovascular disease (Duarte and Lunec 2005). It has been demonstrated that ascorbic acid improves endothelium-dependent and -independent vasodilation in patients with coronary artery disease (Heitzer et al. 2001). A recent study demonstrated that ascorbic acid reduced oxidative stress in testicular tissue of mice exposed to Cd (Acharya et al. 2008). Since there is lack of information about the effect of ascorbic acid on vascular protection in Cd-intoxicated condition, the present study was designed to elucidate whether supplementation of ascorbic acid could alleviate oxidative stress and improve vascular function in an animal experiment of a mouse model of Cd-induced hypertension and vascular dysfunction.
Materials and methods
Animal treatment
Adult male ICR mice weighing 25–30 g were obtained from the Animal Care Unit of the Faculty of Medicine, Khon Kaen University (Khon Kaen, Thailand). The animals were housed in a temperature and humidity controlled room with a 12 h dark:12 h light cycle, free access to drinking water and standard chow diet (Chareon Pokapan Co. Ltd., Thailand). All animal experimental treatment protocols were reviewed and approved by the Animal Ethics Committee of Khon Kaen University. Animals were randomly divided into six groups of 8–10 animals each: group I—normal control, group II—normal control + ascorbic acid 50 mg/kg b.w., group III—normal control + ascorbic acid 100 mg/kg b.w., group IV—Cd control, group V—Cd + ascorbic acid 50 mg/kg b.w., and group VI—Cd + ascorbic acid 100 mg/kg b.w. The normal control group received deionized water (DI) as drinking water whereas the Cd-treated group received drinking water containing CdCl2 (100 mg/l) continuously for 8 weeks. To avoid any further stress that might be introduced to the animals during Cd treatment, antioxidant ascorbic acid dispersed in DI was intragastrically administered (0.1 ml/10 g b.w.) to animals on each alternate day for the same period as the control group. The concentration of Cd and duration of exposure followed a previous study (Thijssen et al. 2007). As previously report in lead-intoxicated mice, the concentration of ascorbic acid used in this study appears to be non-toxic to a small rodent animal, like mouse (Wang et al. 2006).
Determination of body weights and organ weights
Mice body weights were recorded every week for 8 weeks. At the end of experiment, the liver, kidneys, heart and aorta of each animal were quickly excised and weighed. Samples of these organs were collected for measurement of the Cd content. In the separate set of experiments, the aorta of each animal was collected for measurement of O •−2 production.
Assessment of haemodynamics and arterial pressure responses to vasoactive agents
On the last day of experiments, animals were placed in individual metabolic cages for 24 h. Urine samples were collected, then kept and stored at −20°C until analysis for nitrate/nitrite as nitric oxide (NO) oxidative products. Following the urine collection, mice were anaesthetized with an intraperitoneal injection of ketamine:xylazine (100:2.5 mg/kg). A tracheotomy was performed to facilitate respiration. The animal’s body temperature was monitored using a rectal probe and the body temperature was kept constant at 37 ± 2°C by using a heating pad. The right carotid artery was cannulated with polyethylene tubing connected to a pressure transducer for continuously monitoring arterial blood pressure using the Acqknowledge data acquisition and analysis software (Biopac System Inc., California, U.S.A.). Heart rate (HR) was determined by the software from the blood pressure tracing. The left jugular vein was cannulated with polyethylene tubing for infusion of vasoactive agents. After obtaining stable baseline measurements, an endothelium-dependent vasodilator, acetylcholine (ACh; 10 nmol/kg), an endothelium-independent vasodilator, sodium nitroprusside (SNP; 10 nmol/kg), and an alpha sympathomimetic agent, phenylephrine (Phe; 0.03 μmol/kg), were randomly infused intravenously, while blood pressure was continuously monitored. Following the drug infusion, blood pressure was allowed to return to the baseline level and stabilize for at least 5 min. Changes in blood pressure were expressed as percentages of control values obtained immediately before the administration of the test substance (baseline). At the end of the experiment, blood samples were collected from the abdominal aorta for assays of iron status, antioxidant and oxidative stress makers. Subsequently, the aorta was excised rapidly from the animal and used for measurement of O •−2 production.
Biochemical assays
Assay of O •−2 production
O •−2 production was determined by lucigenin-enhanced chemiluminescence method as described previously (Luangaram et al. 2007; Sompamit et al. 2009). In brief, a vessel segment (4 mm in length) was carefully cleaned and incubated in 450 μl oxygenated Krebs-Ringer bicarbonate solution at 37°C for 30 min. The chemiluminescence signal was measured after the addition of lucigenin (30 μM), and counted in a luminometer (Turner Biosystems, 23 CA, U.S.A.).
Assay of nitrate/nitrite
Accumulation of nitrate and nitrite, the oxidative products of NO, was measured in urine samples using a previously described method (Kukongviriyapan et al. 2008). In brief, the nitrate in urine was reduced to nitrite by nitrate reductase, and then the mixture was reacted with Griess solution (4% sulfanilamide in 0.3% NED). The optical absorbance of samples was measured on an enzyme-linked immunosorbent assay (ELISA) plate reader with a filter wavelength of 540 nm (Tecan GmbH., Grodig, Austria). The amount of creatinine in urine samples was measured, and urinary nitrate/nitrite concentration was expressed as nmol/mg creatinine.
Assay of malondialdehyde
Malondialdehyde (MDA) is the main degradative product of lipid peroxidation and is used as an indicator of cellular damage caused by ROS. MDA in plasma, liver, kidney and heart were analyzed by using thiobarbituric acid as previously described (Naowaboot et al. 2009). In brief, 150 μl plasma or tissue samples were reacted with 10% TCA, 5 mM EDTA, 8% SDS, 0.5 μg/ml of BHT and 0.6% TBA. The mixture was boiled in a water bath for 30 min and then cooled, it was centrifuged at 10000×g and 25°C for 5 min. The absorbance of the supernatant was measured at 532 nm by spectrophotometer. The amount of MDA in tissue was calculated using a standard curve of 1,1,3,3-tetra-ethoxypropane (0.3–10 μmol/l). The MDA concentration in the tissues was normalized against the protein concentration. Protein was determined by the Bradford dye binding method.
Assay of protein carbonyl
Protein oxidation in plasma was assessed by the determination of carbonyl groups based on the reaction with dinitrophenyl hydrazine (DNPH) as previously described (Somparn et al. 2007) with modifications (Sompamit et al. 2009). Briefly, plasma samples were incubated with 15 mM DNPH in 3.6 M HCl for 1 h in the dark. Protein was precipitated, washed, and then dissolved in 6 M guanidine. The protein carbonyl content was determined by a spectrophotometric method at a wavelength of 360 nm. The protein amount was assayed by Bradford dye binding method.
Assay of glutathione
Total glutathione (GSH) in the whole blood was assayed by a previously described method (Kukongviriyapan et al. 2008), and glutathione disulfide (GSSG) was analyzed after treating the blood sample with M2VP, a GSH scavenger. Briefly, a 100 μl sample of whole blood was reacted with 10 μl 33 mM M2VP or distilled water, and subsequently treated with 5% cold MPA to precipitate protein. The supernatant obtained after centrifugation was used in the enzymatic coupling assay for GSH by using a spectrophotometer (Biochrom Ltd., Cambridge, UK). The redox ratio was calculated as GSH/GSSG.
Assay of iron status
To investigate whether ascorbic acid modifies the Cd absorption by increased iron absorption, the iron status, including serum iron, serum ferritin, and serum non-transferrin bound iron (NTBI) were assessed in mice exposed to Cd with or without ascorbic acid treatment. The levels of serum iron and ferritin were measured by routine procedure with a Cobas Integra 800 instrument according to the manufacturer’s instructions (Roche/Hitachi Cobas C Systems, Tokyo, Japan). The serum iron assay was based on the release of Fe(III) from transferrin by guanidine hydrochloride. After reduction to Fe(II), the bivalent ions produced a red colored complex with FerroZine. The color intensity was directly proportional to the iron concentration and could be measured photometrically at 570 nm. The serum ferritin assay was based on the immunological agglutination principle with enhancement of the reaction by latex. The precipitate was determined turbidimetrically at 570 nm. The level of NTBI was measured by a previously described method (Jittangprasert et al. 2004) with some modifications. Briefly, an aliquot of 100 μl of serum ultrafiltrate was diluted 1:2 (v/v) with 0.50 M HEPES buffer (pH 7.0). A 25 μl of a reducing agent, 0.15 M sodium thioglycollate (TGA), and 25 μl of 0.05 M bathophenanthrolinedisulfonic acid, disodium salt (BPT), a chromogen for Fe(II), were then added to the solution for colorimetric measurement of the Fe(II)–BPT complex. The solution was then equilibrated for 90 min at room temperature in order for the formation of colored complex to reach equilibrium before measurement of absorbance at 537 nm.
Assay of Cd contents
Samples of blood, liver, kidneys, heart and aorta (thoracic and abdominal aorta) were digested with HNO3 and H2O2 under pressure in a closed vessel. Cd concentrations in all samples were determined as previously described (Evans and Giflio 1993; Morales et al. 2006) by using inductively coupled plasma mass spectrometry (ICP-MS) method (Agilent 7500 ICP-MS model, Santa Clara, CA, USA) according to the manufacturer’s recommendation. The Cd contents were expressed in μg/l and μg/g of tissue wet weight.
Statistical analysis
Data are expressed as means ± SE, and n refers to the number of animals used. Data comparisons were carried out using one-way analysis of variance (ANOVA), followed by post-hoc Duncan’s multiple range test. Statistical significance was assigned at a P value of less than 0.05.
Results
Effects of ascorbic acid on haemodynamics and vascular reactivities
The administration of CdCl2 caused a significant increase in systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean arterial blood pressure (MAP) when compared with control group, but there was no different in HR in all groups (Table 1). Mice treated with ascorbic acid at the study concentrations showed no significant differences in HR, systolic pressure, diastolic pressure, and mean arterial pressure when compared with those receiving the vehicle alone (Table 1). Cd-treated mice that received ascorbic acid (50 or 100 mg/kg) showed a significant reduction in MAP to near control values. Moreover, administration with Cd significantly attenuated the vascular responses to Phe, ACh, and SNP when compared with normal controls as shown in Fig. 1. These results indicate that Cd caused an impairment of vasorelaxation as well as vasoconstriction. The impairment of vascular responses was largely prevented by ascorbic acid (Fig. 1). Ascorbic acid at the high dose could markedly increase vascular responses to Phe about 39 vs. 26%, ACh 46 vs. 30%, and SNP 41 vs. 31% when compared with mice that received Cd alone.

Effect of ascorbic acid on MAP responses to vasoactive agents, including Phe, ACh, and SNP in all experimental groups. Mice received CdCl2 (100 mg/l) alone or combined with ascorbic acid (50 or 100 mg/kg, p.o.). MAP mean arterial pressure. Results are expressed as mean ± SE, n = 8–10/group. * P < 0.05 compared with normal control; # P < 0.05 compared with Cd control
Effects of ascorbic acid on oxidant and antioxidant status
Administration of ascorbic acid (50 or 100 mg/kg) did not change the normal levels of oxidant and antioxidant parameters in control mice when compared with those receiving the vehicle alone (Table 2). Exposure to Cd for 8 weeks induced a marked O •−2 and NO productions as indicated by the amount of O •−2 in thoracic aorta and urinary nitrate/nitrite level when compared with those of normal control group (P < 0.001, Table 2). Ascorbic acid at doses of 50 and 100 mg/kg markedly reduced the rate of O •−2 production and decreased the levels of nitrate/nitrite in comparison to mice treated with Cd alone. Cd increased plasma protein carbonyl and also induced a remarkable increase of MDA levels in plasma, liver, kidney and heart (Table 2). Moreover, administration with ascorbic acid, especially at high dose, significantly reduced the levels of protein carbonyl and MDA in plasma and in all tissues (P < 0.01, Table 2). With regard to the antioxidant status, the blood antioxidant GSH and the redox ratio of GSH/GSSG were dramatically reduced after 8 weeks exposure to Cd, and administration with ascorbic acid significantly increased the level of GSH as well as the redox ratios (P < 0.01, Table 2).
Effect of ascorbic acid on iron status
Treatment of ascorbic acid protected against Cd-induced vascular dysfunction and oxidative stress might be related to interference with Cd absorption by increased iron bioavailability. Serum iron and NTBI levels in plasma were analyzed. There were no significant changes in serum iron in all experimental groups (Table 3). Moreover, serum ferritin and NTBI levels which reflect the iron load in the body were also unaltered. This indicates that ascorbic acid dose not interfere the intestinal uptake of Cd in mice fed with iron sufficient diet.
Effects of ascorbic acid on body weights and organ weights
At the end of experiment, the body weights of animals in all groups were increased as compared to baseline data. Mice in the control group received DI water showed a body weight gain about 39%, which is about 19% more than mice received Cd alone (Table 4). Supplementation of ascorbic acid, especially at high dose significantly improved the body weight gain. Moreover, it revealed that the percent ratios of liver, kidneys and heart weights to body weights were increased in mice treated with Cd, and this toxic effect was partial prevented by ascorbic acid treatment (P < 0.01; Table 4).
Effects of ascorbic acid on Cd accumulations in blood and tissues
The effect of ascorbic acid against Cd accumulations in blood and tissues, including liver, kidneys, heart, aorta and whole blood is shown in Table 5. Cd content in blood and tissues of the normal control mice with or without ascorbic acid treatment was below the level of detection limit. A marked elevation of Cd concentrations in liver, kidneys, heart, aorta, and whole blood was found in mice exposed to Cd (Table 5). Supplementation with ascorbic acid significantly decreased Cd accumulations in blood and the tissues in a dose-dependent manner (P < 0.01, Table 5).
Discussion
A major finding of this study highlighted the beneficial effect of ascorbic acid on subchronic Cd-induced cardiovascular toxicity. Ascorbate improves vascular function by lowering high blood pressure and restoring vascular reactivities to vasoactive substances in mice intoxication with Cd. The plausible mechanisms might be related to the ability of ascorbic acid to suppress oxidant formation and maintain GSH and redox balance.
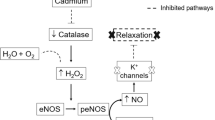
It was found that mice with subchronic exposure to Cd had an increase in blood pressure and attenuation of vascular responsiveness when compared with normal controls, suggesting that vascular dysfunction occurs in these animals. It has been demonstrated that the vascular wall is one of the target organs of Cd deposition, the accumulation of Cd in the aorta facilitates the weakening of the aorta through the adverse effects on smooth muscle cell metabolism (Abu-Hayyeh et al. 2001). Based on our observation, we found an increase in Cd content in the aorta of mice exposed to Cd, suggesting that the attenuation of vascular reactivity to vasoactive agents in this study might be due to the long-term damage of endothelial and VSMCs caused by Cd intoxication. Results from numerous studies suggest that Cd-induced endothelial cell death in vivo significantly alters the endothelial function by increasing endothelial permeability, thereby disrupts the endothelial integrity and contributes to vascular dysfunction (Messner and Bernhard 2010). The blunted response of ACh-induced endothelial vasodilation after Cd exposure can be explained by the inhibitions of endothelial NO synthase (eNOS) phosphorylation (Majumder et al. 2008) and eNOS expression (Yoopan et al. 2008). Moreover, the impairment of Phe-induced contraction may be related to Cd inhibited extracellular Ca2+ independent contractile response by directly disrupting the intracellular signal transduction pathway (Wakabayashi et al. 1995).
The underlying mechanisms of the efficacy of ascorbic acid on Cd-induced hypertension, and vascular dysfunction were investigated in association with the potent antioxidant activity. Cd causes oxidative stress by inducing the generation of ROS, reducing the antioxidant defense systems of cells by depleting GSH, inhibiting SH-dependent enzymes, interfering with some essential metals needed for antioxidant enzyme activities, and/or increasing susceptibility of cells to oxidative attack by altering the membrane integrity and fatty acid composition (Gaubin et al. 2000; Tandon et al. 2003; Messner and Bernhard 2010). Cd exposure is associated with increased production of O •−2 and NO (Hassoun and Stohs 1996; Ramirez and Gimenez 2003). As shown in our results, Cd exposure causes an increase in O •−2 production in the thoracic aorta, and this could effectively scavenge NO to form a strong oxidant peroxynitrite (ONOO−). Treatment with ascorbic acid markedly decreased O •−2 production in aortic tissue and urinary nitrate/nitrite level in a dose-dependent manner. It resulted in reduced formation of peroxynitrite in the endothelial cells and increase bioavailability of NO. It is also suggested that ascorbic acid has a direct effect on reduction of nitrite to NO, and activates of either eNOS or smooth muscle guanylyl cyclase (May 2000). In hypertensive animal study, it revealed that ascorbic acid is critical in normalizing endothelial dysfunction through the regulation of eNOS and NAD(P)H oxidase activities (Ulker et al. 2003).
It seems likely that the free radical scavenging activity of ascorbic acid is well correlated with a decrease in blood pressure of Cd-treated mice. Interestingly, ascorbic acid can donate a hydrogen atom and form a relatively stable ascorbyl free radical. As a scavenger of ROS and RNS, ascorbic acid has been shown to be effective against the O •−2 , H2O2, the OH• and singlet oxygen (Weber et al. 1996). Next, it could reduce reactive free radicals through a formation of the less reactive compound (Padayatty et al. 2003). Ascorbic acid can reduce the initiating ROS so that initial or continued lipid peroxidation is inhibited. The other study supported that the administration of ascorbic acid, vitamin E, and selenium caused a significant decrease in lipid peroxidation levels and an increase in GSH levels in the kidney of rats given Cd (Karabulut-Bulan et al. 2008). It is interesting to find that ascorbic acid dose-dependently increased GSH level and improved GSH/GSSG ratio in the blood cells in mice exposed to Cd alone. Additionally, the protection might be partially due to the elevation of GSH apart from the direct antioxidant effect of ascorbic acid (Seifi et al. 2009). Taken together, the improvement of vascular function and reduction in blood pressure of mice exposed to Cd and received ascorbic acid might considerably contribute to the antioxidant properties of ascorbic acid.
It has been suggested that the gastrointestinal absorption of Cd depends on the levels of iron in the body (Hamilton and Valberg 1974; Flanagan et al. 1978), and this effect is associated with the divalent metal transported I (DMT1) (Park et al. 2002). Since the gastrointestinal absorption of Cd is enhanced in iron-deficient animals, but reduced in iron-replete animals (Ryu et al. 2004; Kim et al. 2007). In our study, we found that the iron status of mice fed with iron sufficient diet remains unchanged after subchronic exposure to Cd. This suggests that supplemented Cd might not interfere with iron absorption in iron-sufficient mice. As ascorbate was shown to increase iron absorption in animals (Van Campen 1972), the increased iron absorption might hypothetically interfere with Cd absorption by competing with DMT1. However, it was turned out that ascorbate did not alter serum iron and NTBI levels in iron-sufficient mice. This does not support the possibility that ascorbate enhanced iron absorption, thereby, reduced body Cd. It is still possible that ascorbate increases turnover rate of Cd by yet to determine the mechanism.
Cd accumulates mainly in the liver and kidney which causes severe tissue damage in these organs. Cd induces production of metallothionein (MT), a low molecular-weight protein that has high affinity for the metal (Klaassen et al. 1999; Nordberg and Nordberg 2000). The amount of MT-bound Cd and non-MT-bound Cd ions may cause hepato- and nephrotoxicity (Nordberg and Nordberg 1987). The large reduction in Cd concentration in the blood after ascorbic acid treatment suggests that ascorbate might interfere with the gastrointestinal absorption of Cd. This is an interesting issue and is needed for further exploration.
In this study, the haemodynamics and vascular responsiveness of Cd-treated mice treated with ascorbic acid were markedly improved to near normal control values whereas the levels of Cd in tissues were partially reduced. However, the Cd-lowering effect of ascorbate may still play some roles in overall beneficial effects, since it is possible that certain threshold levels of Cd in tissues may be necessay to develop vascular dysfunction, a partial reduction of tissue Cd might still be relevant to protecive effects. Interestingly, the cardiovascular benefit of ascorbate is intimately associated with suppression of oxidative stress, i.e., tissue MDA, plasma protein carbonyl and redox status. Our recent work has shown that 2,3-dimercaptosuccinic acid (DMSA), a known metal chelator protected cardiovascular function by metal chelation and inhibition of oxidative stress (Sompamit et al. 2010). The present study suggests that the antioxidative effects of ascorbate play important role in cardiovascular protective effects.
In conclusion, this study demonstrates that Cd-induced vascular dysfunction is associated with increased oxidative stress. Administration of ascorbic acid has a marked protective effect on Cd-induced oxidative stress and vascular dysfunction which seems to be at least based on its strong antioxidant and free radical scavenging properties. Collectively, these findings suggest that dietary supplementation of ascorbic acid may be useful to prevent oxidative stress and vascular dysfunction during Cd intoxication.
References
Abu-Hayyeh S, Sian M, Jones KG, Manuel A, Powell JT (2001) Cadmium accumulation in aortas of smokers. Arterioscler Thromb Vasc Biol 21:863–867
Acharya UR, Mishra M, Patro J, Panda MK (2008) Effect of vitamins C and E on spermatogenesis in mice exposed to cadmium. Reprod Toxicol 25:84–88
Bertin G, Averbeck D (2006) Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie 88:1549–1559
Duarte TL, Lunec J (2005) Review: when is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic Res 39:671–686
Evans H, Giflio J (1993) Interferences in inductivity coupled plasma mass spectrometry. J Anal At Spectrom 8:1–18
Flanagan PR, McLellan JS, Haist J, Cherian G, Chamberlain MJ, Valberg LS (1978) Increased dietary cadmium absorption in mice and human subjects with iron deficiency. Gastroenterology 74:841–846
Fowler BA (2009) Monitoring of human populations for early markers of cadmium toxicity: a review. Toxicol Appl Pharmacol 238:294–300
Gaubin Y, Vaissade F, Croute F, Beau B, Soleilhavoup J, Murat J (2000) Implication of free radicals and glutathione in the mechanism of cadmium-induced expression of stress proteins in the A549 human lung cell-line. Biochim Biophys Acta 1495:4–13
Hamilton DL, Valberg LS (1974) Relationship between cadmium and iron absorption. Am J Physiol 227:1033–1037
Hassoun EA, Stohs SJ (1996) Cadmium-induced production of superoxide anion and nitric oxide, DNA single strand breaks and lactate dehydrogenase leakage in J774A.1 cell cultures. Toxicology 112:219–226
Haytowitz DB (1995) Information from USDA’s Nutrient Data Bank. J Nutr 125:1952–1955
Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T (2001) Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104:2673–2678
IARC (1993) Chemicals, groups of chemicals, complex mixtures, physical and biological agents and exposure circumstances to be evaluated in future
Jarup L, Akesson A (2009) Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol 238:201–208
Jittangprasert P, Wilairat P, Pootrakul P (2004) Comparison of colorimetry and electrothermal atomic absorption spectroscopy for the quantification of non-transferrin bound iron in human sera. Southeast Asian J Trop Med Public Health 35:1039–1044
Karabulut-Bulan O, Bolkent S, Yanardag R, Bilgin-Sokmen B (2008) The role of vitamin C, vitamin E, and selenium on cadmium-induced renal toxicity of rats. Drug Chem Toxicol 31:413–426
Kim DW, Kim KY, Choi BS, Youn P, Ryu DY, Klaassen CD, Park JD (2007) Regulation of metal transporters by dietary iron, and the relationship between body iron levels and cadmium uptake. Arch Toxicol 81:327–334
Kitamura M, Hiramatsu N (2010) The oxidative stress: endoplasmic reticulum stress axis in cadmium toxicity. Biometals. doi:10.1007/s10534-010-9296-2
Klaassen CD, Liu J, Choudhuri S (1999) Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol 39:267–294
Kukongviriyapan V, Somparn N, Senggunprai L, Prawan A, Kukongviriyapan U, Jetsrisuparb A (2008) Endothelial dysfunction and oxidant status in pediatric patients with hemoglobin E-beta thalassemia. Pediatr Cardiol 29:130–135
Luangaram S, Kukongviriyapan U, Pakdeechote P, Kukongviriyapan V, Pannangpetch P (2007) Protective effects of quercetin against phenylhydrazine-induced vascular dysfunction and oxidative stress in rats. Food Chem Toxicol 45:448–455
Majumder S, Muley A, Kolluru GK, Saurabh S, Tamilarasan KP, Chandrasekhar S, Reddy HB, Purohit S, Chatterjee S (2008) Cadmium reduces nitric oxide production by impairing phosphorylation of endothelial nitric oxide synthase. Biochem Cell Biol 86:1–10
Mandl J, Szarka A, Banhegyi G (2009) Vitamin C: update on physiology and pharmacology. Br J Pharmacol 157:1097–1110
May JM (2000) How does ascorbic acid prevent endothelial dysfunction? Free Radic Biol Med 28:1421–1429
Messner B, Bernhard D (2010) Cadmium and cardiovascular diseases: cell biology, pathophysiology, and epidemiological relevance. Biometals. doi:10.1007/s10534-010-9314-4
Morales AI, Vicente-Sanchez C, Sandoval JM, Egido J, Mayoral P, Arevalo MA, Fernandez-Tagarro M, Lopez-Novoa JM, Perez-Barriocanal F (2006) Protective effect of quercetin on experimental chronic cadmium nephrotoxicity in rats is based on its antioxidant properties. Food Chem Toxicol 44:2092–2100
Nakagawa H, Nishijo M (1996) Environmental cadmium exposure, hypertension and cardiovascular risk. J Cardiovasc Risk 3:11–17
Naowaboot J, Pannangpetch P, Kukongviriyapan V, Kukongviriyapan U, Nakmareong S, Itharat A (2009) Mulberry leaf extract restores arterial pressure in streptozotocin-induced chronic diabetic rats. Nutr Res 29:602–608
Nordberg M, Nordberg GF (1987) On the role of metallothionein in cadmium induced renal toxicity. Experientia Suppl 52:669–675
Nordberg M, Nordberg GF (2000) Toxicological aspects of metallothionein. Cell Mol Biol (Noisy-le-grand) 46:451–463
Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee JH, Chen S, Corpe C, Dutta A, Dutta SK, Levine M (2003) Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr 22:18–35
Park JD, Cherrington NJ, Klaassen CD (2002) Intestinal absorption of cadmium is associated with divalent metal transporter 1 in rats. Toxicol Sci 68:288–294
Prozialeck WC, Edwards JR, Woods JM (2006) The vascular endothelium as a target of cadmium toxicity. Life Sci 79:1493–1506
Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD (2008) The vascular system as a target of metal toxicity. Toxicol Sci 102:207–218
Ramirez DC, Gimenez MS (2003) Induction of redox changes, inducible nitric oxide synthase and cyclooxygenase-2 by chronic cadmium exposure in mouse peritoneal macrophages. Toxicol Lett 145:121–132
Ryu DY, Lee SJ, Park DW, Choi BS, Klaassen CD, Park JD (2004) Dietary iron regulates intestinal cadmium absorption through iron transporters in rats. Toxicol Lett 152:19–25
Satarug S, Moore MR (2004) Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect 112:1099–1103
Satarug S, Nishijo M, Ujjin P, Vanavanitkun Y, Moore MR (2005) Cadmium-induced nephropathy in the development of high blood pressure. Toxicol Lett 157:57–68
Satarug S, Garrett SH, Sens MA, Sens DA (2010) Cadmium, environmental exposure, and health outcomes. Environ Health Perspect 118:182–190
Seifi B, Kadkhodaee M, Karimian SM, Zahmatkesh M, Shams S, Bakhshi E (2009) Reduction of kidney damage by supplementation of vitamins C and e in rats with deoxycorticosterone-salt-induced hypertension. Iran J Kidney Dis 3:197–202
Sompamit K, Kukongviriyapan U, Nakmareong S, Pannangpetch P, Kukongviriyapan V (2009) Curcumin improves vascular function and alleviates oxidative stress in non-lethal lipopolysaccharide-induced endotoxaemia in mice. Eur J Pharmacol 616:192–199
Sompamit K, Kukongviriyapan U, Donpunha W, Nakmareong S, Kukongviriyapan V (2010) Reversal of cadmium-induced vascular dysfunction and oxidative stress by meso-2,3-dimercaptosuccinic acid in mice. Toxicol Lett 198:77–82
Somparn N, Kukongviriyapan U, Tassaneeyakul W, Jetsrisuparb A, Kukongviriyapan V (2007) Modification of CYP2E1 and CYP3A4 activities in haemoglobin E-beta thalassemia patients. Eur J Clin Pharmacol 63:43–50
Tandon SK, Singh S, Prasad S, Khandekar K, Dwivedi VK, Chatterjee M, Mathur N (2003) Reversal of cadmium induced oxidative stress by chelating agent, antioxidant or their combination in rat. Toxicol Lett 145:211–217
Thevenod F (2010) Catch me if you can! Novel aspects of cadmium transport in mammalian cells. Biometals. doi:10.1007/s10534-010-9309-1
Thijssen S, Maringwa J, Faes C, Lambrichts I, Van Kerkhove E (2007) Chronic exposure of mice to environmentally relevant, low doses of cadmium leads to early renal damage, not predicted by blood or urine cadmium levels. Toxicology 229:145–156
Ulker S, McKeown PP, Bayraktutan U (2003) Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension 41:534–539
Van Campen D (1972) Effect of histidine and ascorbic acid on the absorption and retention of 59 Fe by iron-depleted rats. J Nutr 102:165–170
Wakabayashi I, Sakamoto K, Hatake K (1995) Inhibitory effects of cadmium ion on extracellular Ca(2+)-independent contraction of rat aorta. Eur J Pharmacol 293:133–140
Wang Y, Fang J, Leonard SS, Rao KM (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36:1434–1443
Wang C, Zhang Y, Liang J, Shan G, Wang Y, Shi Q (2006) Impacts of ascorbic acid and thiamine supplementation at different concentrations on lead toxicity in testis. Clin Chim Acta 370:82–88
Weber P, Bendich A, Schalch W (1996) Vitamin C and human health—a review of recent data relevant to human requirements. Int J Vitam Nutr Res 66:19–30
WHO (1993) Evaluation of certain food additives and contaminants. Forty-first Report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series 837
Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M (2008) Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem 283:4252–4260
Yoopan N, Watcharasit P, Wongsawatkul O, Piyachaturawat P, Satayavivad J (2008) Attenuation of eNOS expression in cadmium-induced hypertensive rats. Toxicol Lett 176:157–161
Acknowledgments
This work was supported by the Faculty of Medicine, and the Graduate School Research Funds, Khon Kaen University, Thailand. Wanida Donpunha was supported by a Ph.D. scholarship from Graduate School, Khon Kaen University, Thailand.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Donpunha, W., Kukongviriyapan, U., Sompamit, K. et al. Protective effect of ascorbic acid on cadmium-induced hypertension and vascular dysfunction in mice. Biometals 24, 105–115 (2011). https://doi.org/10.1007/s10534-010-9379-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-010-9379-0