
Abstract
Adsorption of dissolved organic compounds onto mineral surfaces is increasingly recognized as a significant, if not dominant, carbon stabilisation mechanism in many soils. By utilising carbon-13 enriched dissolved organic carbon (DOC) source materials in a repeated leaching-sorption-incubation study, we show here that the biochemical composition of mineral-retained organic matter (OM) is similar across four different classes of clay minerals but the quantity and stability of this OM is both a function of source material and clay mineralogy. Three to eight times as much carbon was retained on a mass basis when the same amount of DOC derived from eucalyptus versus wheat litter was applied, and the retained wheat-derived OM was up to 2.4 times more degradable than that of the eucalyptus source. For both litter types, carbon retention across the clay types was not significantly different; whereas, the stability of the retained OM was different but depended on which litter extract had been applied. The wheat-derived DOC was more stable when retained by allophane and oxides than by illite and smectite. Solid-state 13C NMR spectroscopic results indicated that despite large compositional differences in both source litter and resultant DOC, the composition of the mineral-retained OM was similar across clay classes with lignin-derived aromatic and carboxylic compounds dominating. Differences in the amount of carbon retained were related to differences in the proportions of aromatic, phenolic and carboxylic C in the DOC produced from the two litter sources. Differences in the stability across the clay classes were correlated with the abundance of metals and short-range ordered minerals. These results suggest that whenever reactive mineral surfaces and metals are present in a soil, a similar form of relatively unaltered litter derived OM can be adsorbed but that the longer term stability of sorbed OM, and thus in situ composition, will be a function of the mineralogy (reactivity) of the specific minerals involved in the binding process.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Understanding the mechanisms of soil organic matter (SOM) stabilisation and destabilisation is a basic requirement for furthering our understanding of the role organic matter turnover plays in plant nutrition and building SOM stocks as an effective greenhouse gas mitigation strategy (Smernik and Skjemstad 2009). The traditional concept (e.g. Stevenson 1982) of a highly condensed macromolecular structure for stable SOM, often termed humic substances, and even the exclusion of simple recognizable biomolecules from the stable SOM pool have been thoroughly challenged in recent years (Kleber et al. 2007; Piccolo 2001; Sutton and Sposito 2005). With the aid of advanced molecular techniques, a new view (Schmidt et al. 2011) has emerged where humic substances are thought to be dynamic supramolecular associations (Kleber et al. 2007; Piccolo 2001) of diverse, relatively low molecular weight components. These components include recognizable but often partially-oxidized biomolecules (Baldock et al. 2004; Schnitzer 2000), stabilised by numerous mechanisms, with hydrophobic interactions and hydrogen bonding being of particular importance (Baldock and Skjemstad 2000; Schnitzer 2000; Sutton and Sposito 2005). Simple relatively fresh and potentially labile biomolecules can contribute directly to this stable SOM pool via retention on mineral surfaces (Kramer et al. 2012) and there does not necessarily have to be a long slow aging process to produce stable humus (Kleber et al. 2007). Sorption of relatively low molecular weight dissolved organic compounds onto mineral surfaces in many soils may be a critical mechanism for carbon accumulation and long-term stabilisation against decomposition (Kögel-Knabner et al. 2008; Kaiser and Kalbitz 2012).
Dissolved organic carbon (DOC) leaching into minerals soils often represents only a small fraction of annual net primary production (Kindler et al. 2011), yet the contribution of DOC retention to long-term soil carbon storage has been estimated to range from 9 to 89 % of total mineral soil carbon stocks in a diverse range of ecosystems (Kalbitz and Kaiser 2008; Michalzik et al. 2003; Neff and Asner 2001; Sanderman and Amundson 2009). For example, Sanderman and Amundson (2009) combined field based measurements of DOC fluxes with radiocarbon measurements of both DOC and SOC to estimate that 9 and 20 % of total SOC can be attributed to DOC sorption in a grassland and forested ecosystem, respectively. Others have scaled laboratory based measurement of sorption potential with decomposition rates of OC in solution and after sorption to various mineral surfaces (e.g. Kalbitz and Kaiser 2008; Michalzik et al. 2003) or incorporated these types of results into ecosystem carbon models (e.g. Neff and Asner 2001). Overall, these studies all suggest the prominent role of organo-mineral associations in long-term carbon stabilisation.
The large variability in the above estimates suggests that some soil types are more effective in stabilising carbon via mineral interactions. While some of the variability is surely due to methodological differences (e.g. field- or lab-based measurements, use of fresh or pedogenically-derived clay minerals) and climate driven differences in soil water fluxes, at a micro-scale differences in clay mineralogy and the soil solution environment are major drivers controlling both the magnitude and stability of organo-mineral associations. Interactions between OM and mineral surfaces can be grouped into coulombic (i.e. electrostatic) and non-coulombic (i.e. Van der Waals forces) interactions (Feng et al. 2005 and Mikutta et al. 2007 provide excellent overviews). In many soils, the direct exchange of an organic functional group with a hydroxyl or water group on a mineral surface (i.e. ligand exchange) has been found to be the dominant and most stable form of organo-mineral association (Gu et al. 1994; Kögel-Knabner et al. 2008; Mikutta et al. 2007). Cation bridging of OM, often via a carboxylic functional group, to permanently negatively charged mineral surfaces represents a second important binding mechanism operative in many soils (Arnarson and Keil 2000; Mikutta et al. 2007). Sutton and Sposito (2006) stressed the importance of polar organic anions forming associations with the hydration shell of metals (i.e. water bridging) as an important OM retention mechanism. Keiluweit and Kleber (2009) suggested that dominantly apolar aromatic rings can directly bind to mineral surfaces via cation-pi and pi–pi electron donor–acceptor interactions. Importantly, these different binding mechanisms suggest differing degrees of selectivity towards particular classes of organic compounds, bioavailability when sorbed, and desorbability, resulting in potentially large differences in the importance of mineral stabilisation to long-term carbon storage.
Precipitates and condensates of OM with polyvalent cations (Ca2+, Fe3+, Al3+) represent another important class of interactions which will remove carbon from solution (Kalbitz and Kaiser 2008; Römkens and Dolfing 1998; Scheel et al. 2007, 2008). As with most of the coulombic interactions with mineral surfaces mentioned above, there appears to be a strong preference for aromatic and carboxylic C compounds in these precipitates (Scheel et al. 2008). Likely because of the numerous inter- and intra-molecular linkages within the OM and with the polyvalent cations, the bioavailability of these precipitates is also often reduced relative to the bioavailability in a free solution (Scheel et al. 2007) and potentially more so than that of surfically bound OM (Kalbitz and Kaiser 2008). Given that it is often difficult to separate DOC retention via binding to mineral surfaces versus precipitation of organo-metal complexes onto mineral surfaces, we adopt the terminology of mineral-retained OM to refer to the combined actions of both processes.
As the discussion of binding mechanisms above suggests, different mineral classes will retain OM differentially. Oxides and hydroxides of Fe and Al, present as either crystalline minerals or associated with other mineral particles, typically retain OM strongly due to a dominance of ligand exchange interactions (Wershaw et al. 1996; Qualls 2000). Short-range ordered (SRO) non-crystalline minerals (e.g. allophane, ferrihydrite) with extremely high surface areas and porosity have been shown to be particularly effective in retaining OM (Kramer et al. 2012; Mikutta et al. 2009) most likely due to a combination of retention mechanisms. Chorover et al. (2004) suggested that strong inner-sphere complexation reactions at exposed metal centers were particularly important in soils with SRO minerals. Retention of OM via cation bridging to 2:1 phyllosillicate clays with permanent negative charge will select for organic compounds with negative charges; however, many if not most organic compounds found in soil solution can have regions of net negative charge (Kleber et al. 2007). While shrink/swell 2:1 phyllosillicates, such as found in the smectite group, have greater cation exchange capacity (CEC) and surface area than non-expanding 2:1 phyllosillicate clays (i.e. illite) suggesting a greater stabilization potential, it is unclear whether this additional binding capacity would affect OM-mineral interactions due to the narrow diameter of the interlayer space. In fact, Theng et al. (1986) found that intercalation of OM could only occur in acidic soils (pH < 4) and methods for quantifying intercalated OM are highly unreliable (Leifeld and Kögel-Knabner 2001). Additionally, the electrostatic forces involved in cation/water bridging are weaker than for ligand exchange suggesting that this OM will be more desorbable and bioavailable. Finally, in most soil solutions polyvalent cations, especially Al and Fe, are present either in free solution or loosely associated with various mineral surfaces. In either case, these cations can promote precipitation of OM out of solution and further stabilize already mineral-retained OM as described earlier (i.e. Scheel et al. 2007).
In the present study, a two factorial (litter and clay type) repeated sorption-incubation experiment using 13C-enriched litter was performed that quantified (1) the amount of new DOC retained to mineral surfaces, (2) the stability of this retained DOC, and (3) the biochemical composition of the mineral-retained DOC. Eucalyptus and wheat litter sources were chosen to represent a large biochemical contrast in source material for the DOC. Despite large initial differences in litter composition, prior observations in natural systems (Sanderman and Kramer 2013) suggested that the resulting leachates will at least partially converge with respect to biochemical composition resulting in only minor differences in the behaviour of the two leachates when applied to the different clay types. Four clay fractions representing contrasting mineralogy (short-range ordered allophane, fine grained illite, smectite, and hydroxides of Fe and Al) were isolated from field soils. The diversity of these minerals provided a potential for significant differences in retention and stabilisation of OC found in the two litter leachates when applied over the course of 11 weeks. The use of 13C-enriched litter allowed the biochemical composition of the mineral-retained DOC to be determined via solid-state 13C NMR spectroscopy with high precision.
Methods
Experimental design
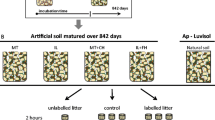
A two-step controlled incubation-leaching-sorption experiment was conducted over an 11 week period (Fig. 1). At t0, 15 g d.w. of each of the two 13C-enriched litter sources (see “Litter material” section) was incubated in quadruplicate in the dark at 25 °C after initial wetting. After this 1 week pre-incubation period, water-soluble OM was extracted on a weekly basis from each replicate by mixing the litter with 250 ml of 0.01 M CaCl2 solution. After a 1 h equilibration time, the litter extracts were then filtered through a 0.7 μm glass fiber filter (Whatman GF/D), combined into a single composite extract for each litter type and diluted to a final volume of 1.6 l that would then be applied to the four clay treatments. The litter samples were then either placed back into the incubators (odd number weeks) or into 2.25 l air-tight container fitted with septa for gas sampling and then back into the incubators (even number weeks). The length of time that the jars were sealed before collection of a headspace sample for determination of CO2 concentration and isotopic composition varied from 2 to 7 days depending on microbial activity (2 days initially and increasing as respiration rates decreased).

Experimental design. In steps 1–5, litter samples are extracted weekly, incubated and respiration rates determined on alternating weeks. The extracts are then applied to the five clay mixture treatments in the same fashion as demonstrated for illite (steps 6–10). This procedure was repeated for 10 consecutive weeks
The composite extracts from each of the two litter types were applied to the four different clay types (see “Clay material” section) along with a sand-only control in a batch sorption style experiment. Each litter × clay combination (10 in total) was replicated three times. In each replicate, 21 g of the particular clay mixture was split evenly between three 50 ml centrifuge tubes. Immediately following each of the weekly litter extractions, the composite extract solutions were added to the clay treatments in a 6:1 solution-to-solid gravimetric ratio. The tubes were then placed on a reciprocating shaker table for 2 h. After this equilibration period, the samples were centrifuged (10 min at 2,000 rpm), decanted and filtered through a 0.7 μm filter. The filtrates from the three tubes for each replicate were combined to form a single 90 ml sample for further analysis. Upon completion of the batch adsorption, all samples were returned to the incubator. On even numbered weeks, each replicate litter × clay combination (3 centrifuges tubes containing 21 g of material in total) was placed in a 1 l air-tight jar fitted with septa for headspace gas sampling. CO2 was allowed to accumulate for 7 days for the clay mixture incubations before headspace sampling.
While these experimental conditions do not mimic what would happen in natural field soils, this design differs from one-off water extractions of litter and batch sorption experiments with fresh clays in that temporal changes due to microbial activity and, potentially, surface loading on the clays will be captured. The primary goal of this experimental design was to load the different clays with enough 13C-labelled OM to obtain well resolved NMR spectra without the need for any pretreatment which can preferentially lose the mineral-retained OM fraction (Eusterhues et al. 2003).
Litter material
Two types of aboveground litter sources were used in this study: Eucalyptus leaves and wheat straw. These litters were produced with a bulk 13C fraction of 21.5 %, where 13C fraction is the preferred nomenclature for tracer studies (Coplen 2011), by growing the plants in a growth chamber continuously fed with 13C enriched CO2 over a 4 month period (for details of the growth chamber see Butler and Eickhoff 1979).
Clay material
Four soils with contrasting dominant clay mineralogy were chosen for this study (Table 1): (1) a subsurface horizon of an Andosol (Mt Schank, South Australia) rich in short-range ordered non-crystalline minerals; (2) the surface horizon of a tropical Ferralsol (Tallagalla, Queensland) dominated by Fe- and Al-oxides; (3) a subsurface horizon of a Solonetz (Willalooka, South Australia) dominated by illite clays; and (4) the surface horizon of a Vertisol (Toowoomba, Queensland) rich in smectite. The clay fraction (<2 μm) in each of these soils was concentrated by sedimentation. Carbonates, if present, were then removed by treatment with 5 % w/v H2SO3 until visible effervescence ceased. To both maximize potential sorption and signal of newly retained OM during this experiment, an alkaline extraction was used to remove as much organic C as possible. Based on results of Mikutta et al. (2005), we chose a sequential extraction procedure using 1 M NaOCl adjusted to a pH of 8.0 (Kaiser et al. 2002). Upon completion of the extraction procedure, the clays were thoroughly rinsed with milliQ water to remove excess salts, then with a 0.2 M CaCl2 solution to promote flocculation, and finally lyophilized until further use. The dry clays were mixed with acid-washed fine grained quartz sand to achieve a final clay content of 30 % by mass for each of the treatments. Throughout this paper, we name each treatment by its dominant clay mineralogy: allophane, illite, oxide or smectite.
Mineralogy of clay samples was determined by qualitative X-ray diffraction analysis (Tucker 1988). XRD patterns were collected on a PANalytical X’pert Pro MDP system using Fe filtered Co Kα radiation and X’Celerator Si strip X-ray detector with an automatic divergence slit. The step size of the patterns was 0.0167° and the total run time for each pattern was 30 min. Patterns were interpreted using in-house software. Cation exchange capacity of each clay mixture was determined in triplicate by compulsive exchange with ammonium acetate (Chapman 1965). Sodium pyrophosphate (an estimate of organically complexed metals), dithionite-sodium citrate (an estimate of free sesquioxides), and acid ammonium oxalate (an estimate of short-range ordered + organically complexed metals) extractable Al and Fe were also measured in triplicate following the methods outlined in Sheldrick (1984). Concentrations of Al and Fe in extracting solutions were determined by on a Spectro ARCOS ICP-OES (Spectro Analytical Instruments, Germany).
Analytical procedures
Aqueous samples from the weekly litter extracts (n = 1 for each litter at each sampling date) and from the filtrates following batch equilibration of each litter extract with each clay type (n = 3 for each litter × clay combination, 10 combinations in all) were analysed for DOC concentration, 13C enrichment, pH and specific UV adsorption (SUVA).
The concentration and 13C fraction of DOC was determined by isotope ratio mass spectrometry. A Thermalox TOC/TN analyser (Analytical Sciences Ltd., UK) first converted acidified samples into CO2 via high temperature oxidation. The evolved CO2 was then fed through a Cyroprep system using ultrapure He as a carrier gas to concentrate the CO2 before analysis on a Sercon GEO 20/20 IRMS (Sercon Ltd., UK) modified with a low resistance amplifier on the mass 45 Faraday collector to accommodate the highly enriched nature of the samples in this study. See De Troyer et al. (2010) for a full description of the hyphenated setup. Two sets of standards were prepared by dissolving mixtures of glycine and 99 % 13C-labelled glycine-2 (Sigma-Aldrich) in milliQ water: (1) a series of DOC concentration linearity standards prepared at a 13C fraction of 23 % spanning a concentration range from 4 to 200 mg C L−1; and (2) a series of 13C linearity standards prepared at 50 mg C L−1 concentration spanning a 13C fraction range from 1.1 (i.e. natural abundance) to 25 %. Ten percent of samples were collected and analysed in duplicate. The mean error (1 s.d.) of duplicate samples was 6.4 mg C L−1 for DOC concentration and 0.39 % for 13C fraction.
Ultraviolet adsorption (λ = 254 nm) was determined on a Shimadzu 1601 UV/Vis spectrophotometer using a 1 cm quartz cell. The UV adsorption normalized to DOC concentration, or specific UV adsorption (SUVA, l mg C−1 m−1), is used here as a proxy for the abundance of aromatic compounds in the sample (Weishaar et al. 2003). The pH of the extracts and filtrates from even numbered weeks were measured using on a Denver Instrument model 25 pH meter. Headspace CO2 samples from both the litter (n = 4 for each litter) and clay incubations (n = 3 for each litter × clay combination) were analysed for CO2 concentration and 13C content using the same Sercon Geo 20/20 IRMS interfaced with a Gilson gas bench autosampler and Cryoprep. Standards were prepared by acidifying a mixtures of NaCO3 and 99 % 13C-labelled NaCO3 (Sigma-Aldrich) to obtain CO2 enriched at a 13C fraction of 2, 5, 10 and 25 % with a concentration of 2,000 ppm. Linearity standards for calculating CO2 concentration were prepared at 10 % 13C fraction with concentrations ranging from 500 to 5,000 ppm. Ten percent of samples were collected and analysed in duplicate. The mean error (1 s.d.) of duplicate samples was 162 ppm and 0.25 % 13C fraction.
The carbon content and isotopic composition in the initial and final clay mixtures and in the lyophilized DOC samples was also determined by isotope ratio mass spectrometry using a Europa Solid/Liquid elemental analyser interfaced with the same Sercon GEO 20/20 IRMS.
Solid-state 13C cross polarization magic angle spinning (CP/MAS) NMR spectroscopy was used to quantify the chemical composition of carbon in the OM found in the initial and final litter samples, the litter leachates at three occasions (week 1, 5 and 9), the filtrates following batch equilibration with the clay mixtures at week 5, and of the clay mixtures before and after the experiment. Paramagnetic interferences prevented acquisition of NMR spectra on the oxide treatment. Leachate and filtrate samples were frozen and then lyophilized for the solid-state analysis. Clay samples were prepared by washing twice with milliQ water and then separating out the sand by sedimentation. Spectra were acquired on a Bruker 200 Avance spectrometer equipped with a 4.7 T wide-bore superconducting magnet operating at a resonance frequency of 50.33 MHz using a 3.2 μs 195 w 90° pulse with a contact time of 1 ms and a recycle delay of 1 s. Full details of operating conditions can be found in Baldock et al. (2013). The number of scans varied from 200 for litter and high-C DOC samples to 60,000 for the clay mixtures. Line broadening at 50 Hz was only applied to the spectra obtained from the clay mixtures, all other spectra were of extremely high quality due to the abundance of 13C nuclei and no line broadening was needed. Chemical shift values were calculated based on the methyl resonance of hexamethylbenzene at 17.36 ppm. Distribution of signal intensity was then assigned to eight major chemical shift (ppm) regions: 0–45 (Alkyl-C), 45-60 (N-Alkyl/Methoxyl), 60-95 (O-Alkyl), 95-110 (Di-O-Alkyl), 110-145 (Aryl), 145-165 (O-Aryl), 165-190 (Amide/Carboxyl) and 190–210 (Ketone). To aid in the interpretation of the NMR spectra, we applied a five-component molecular mixing model (Baldock et al. 2004) that partitions the sample spectra into estimated contributions from carbohydrate, protein, lignin, lipid and carbonyl C. The mixing model was run without using the C/N ratio as an additional constraint.
Data analysis
The percent retention of new DOC during the sorption experiments was estimated as the difference between the input (i) and post-sorption filtrate (f) concentration of DOC adjusted for desorption of older SOM (DOC old ):
where DOC i and DOC f are the measured concentrations of DOC in the input and filtrate solutions, respectively. DOC old was calculated by multiplying DOC f by the fraction of DOC f that was contributed from the native SOM (f SOM ) using a two-member isotope mixing model:
where the 13C data, reported as 13C fraction (%), for the native SOM was 1.1 % and the input DOC solution averaged 21.5 %. The fraction of new litter derived C present in the filtrate solution is then simply 1 − f SOM .
Carbon dioxide production from the clay treatments with DOC applied was quantified from the build up in CO2 over the 1 week incubation periods and partitioned into respiration of litter-derived DOC and from native SOM still present on the clays using a two-member isotope mixing model (Eq. 2). Due to the nature of the repeated batch sorption experiments, a small amount (~2 ml) of non-mineral associated DOC remained in the pore water after each treatment. We assumed the respiration from the control treatment represented the decomposition of this free DOC and subtracted this amount of respiration away from the total respiration of each of the clay treatments (termed background respiration). The bioavailability of the mineral retained C was then assessed by calculating the percent of retained C that was then respired during the incubation period after adjusting for background respiration. Stability is simply the inverse of bioavailability.
Two-way analysis of variance was performed on the total amount of DOC retained and the percent of mineral-retained DOC that was respired. A repeat measures ANOVA was used to analyze the pH data. All data were tested for normality and equal variance. When appropriate, pairwise comparisons within each litter treatment were made across clay treatments using the conservative Holm–Sidak method. Potential mineralogical controls on both the retention of DOC and the subsequent stability of the mineral-retained DOC were assessed by calculating the Pearson product moment correlation coefficients between each of these C dynamic measurements and the measured mineral properties (i.e., CEC and the various extractable Fe and Al measurements). Analysis of variance and correlation analyses were performed using SigmaPlot version 12 (Systat Software Inc., USA).
To further aid in the interpretation of NMR data, a principal components analysis (PCA) was performed on the distribution of NMR signal intensities across eight major chemical shift regions as defined in Sanderman et al. (2008) using the Primer version 6 software package (PRIMER-E, UK). A hierarchical cluster analysis using the SIMPROF test for significant groupings was also performed on the same data after computing the resemblance matrix using Euclidean distances between samples (Clark 1993).
Results
Litter leaching-incubations
Production of CO2 dropped exponentially in both litter types (Fig. 2a) with mean (±1 SEM (n = 4)) cumulative production being similar at 61.7 ± 9.5 and 60.9 ± 12.6 mg C g C−1 for the Eucalyptus and wheat litters, respectively. The 13C fraction of the respired CO2 decreased more or less linearly from 23.3 to 19.5 % for both litters during the course of the experiment (Fig. 2a).

Temporal decomposition dynamics of eucalyptus (filled symbols) and wheat (open symbols) litters: a CO2 production and C isotopic composition of respired CO2; and b DOC production and associated SUVA and C isotope values of DOC. Error bars for (a) = 1 SEM (n = 4)
Total extraction of carbon from the litters as DOC was also similar between litter types, with 73.3 and 76.0 mg C g C−1 being extracted from the Eucalyptus and wheat litter, respectively. The 13C fraction of the DOC remained steady at 21.5 % for the Eucalyptus litter but the wheat-derived DOC decreased to 19 % before increasing slightly in the last 3 weeks (Fig. 2b).
The chemistry, as determined by solid-state 13C NMR spectroscopy, of the original litter and the residue remaining at after 12 weeks of incubation varied between the two litters (Fig. 3a, b). The original wheat litter was dominated by O-Alkyl and di-O-Alkyl C indicative of cellulose. Smaller amounts of Alkyl and Carbonyl/Amide C were evident and with minor amounts of Aryl and O-Aryl C. In the Eucalyptus litter, signal intensity associated with Alkyl, N-Alkyl/Methoxyl, Aryl, O-Aryl and Carbonyl/Amide C made a more substantial contribution than observed for the wheat litter. Additionally, there was a shift in chemistry from O-Alkyl dominance to Alkyl-C dominance over the course of the incubation for the eucalyptus residues but little change in overall chemistry was noted for the wheat residues (Fig. 3a, b).

Solid-state 13C NMR spectra of litter samples before and after 3 month incubation (a, b), and of DOC in extracts at beginning, middle and end of the incubation experiment (c, d)
Extractable DOC from the two litter sources also varied in chemical character and although little shift in C chemistry was noted for the wheat residue, a substantial decrease in O-Alkyl C was observed in the eucalyptus extracts throughout the course of the experiment (Fig. 3c, d). The combined signal from Aryl and O-Aryl C types averaged 33 % in the eucalyptus but only 20 % in the wheat leachates (see supplemental material for full distribution of signal intensity to chemical shift regions). The SUVA data obtained on the extracts were consistent with the NMR derived variations in Aryl and O-Aryl C content (linear regression of Aryl + O-Aryl C versus SUVA: R2 = 0.88, p = 0.006, n = 6), with the eucalyptus leachates having SUVA values 3–4 times higher than that of the wheat and SUVA values increasing over time for extracts from both litters (Fig. 2b). There were also large differences in the pH of the two leachates, averaging 3.72 ± 0.27 and 6.73 ± 0.15 for eucalyptus and wheat extracts, respectively (Fig. 4).

Mean pH of extracts and filtrates. Error bars = 1 SEM (n = 5). Different letters above bars for each litter type indicate significant differences (α < 0.05)
Sorption dynamics
The percent of the weekly application of Eucalyptus DOC retained increased throughout the course of the experiment for all clay types (Fig. 5a); whereas the weekly wheat DOC retention peaked at week 4 and then declined throughout the remainder of the experiment (Fig. 5b). For both litter types, the isotopic enrichment of the post-sorption filtrate decreased indicating that native SOC was released into solution (Fig. 5c, d).

Temporal changes in percent retention of litter extracts when applied to the four clay treatments (a, b) and in the isotopic composition of filtrates following each DOC application (c, d). The right-hand axis on plots c and d converts the 13C fraction values to percent new carbon (i.e. c derived from the litter sources) in the filtrates using Eq. 2. Data represent mean of three replicates for each treatment
The total amount of new C retained during the experiment varied significantly between litter types (F = 51.43, p < 0.001) but not between clay treatments (F = 2.15, p = 0.13). A total of 276 and 258 mg C was applied as DOC from the eucalyptus and wheat DOC, respectively. Despite these similar input amounts, 3–8 times as much C was retained from the eucalyptus DOC (Fig. 6).

Carbon balance at end of experiment. Whole bars indicate total DOC retained during the experiment (calculated from sorption data). Solid bars indicate amount still present at end of experiment after subtracting respiratory losses: (1) hashed segments indicate litter-derived mineral retained DOC that was retained but then respired; and (2) the white segments represent background respiration of litter-derived DOC that remained in solution. The percent of newly retained C that was respired during the course of the experiment is also given on each bar. Error bars = 1 SEM (n = 3), propagated through all measurements
The stability of the retained DOC varied as a function of both litter (F = 258, p < 0.001) and clay type (F = 166, p < 0.001); however, a significant interaction term (F = 64.1, p < 0.001) precludes interpretation of the main effects (data not shown). For the eucalyptus DOC, there was little variation in the stability of the mineral-retained DOC with only the allophane clay mixture invoking a greater stability than the other three types of clay. However, the allophane and oxide dominated clay mixtures stabilized significantly more of the wheat-derived DOC than the illite and smectite clay mixtures. The sorptive capacity of the oxide treatment may have been underestimated in this study due to NaOCl not being as effective in removing organic matter from Fe oxides (Table 1).
Solid-state NMR spectroscopy results demonstrated that the soil core filtrates were always very similar in chemical composition for each DOC extract type regardless of clay mineralogy (Fig. 7). The filtrates measured at the mid-point of the experiment were all depleted in Aryl and O-Aryl C relative to the input solution and to the chemistry of the mineral retained C (Figs. 7, 8 and supplemental material). This shift in NMR composition from extract to filtrate was more pronounced for the eucalyptus DOC than for the wheat. The molecular mixing model results suggested that the relative proportion of carbohydrate-like C (O-Alkyl C) did not change between the input solution and the filtrates but the filtrates were heavily enriched in proteinaceous compounds (Fig. 9). Figure 9 also demonstrated that while the mineral-retained OM was depleted in carbohydrate C relative to the input solution, 13–19 % of the retained C was identifiable as carbohydrates. Proteins were preferentially left in solution (Fig. 9). In Fig. 8, NMR spectra were also acquired for the initial clay mixtures to demonstrate that the signal we see is due to the 13C-enriched DOC that has been retained on the mineral surfaces and not the native SOM that wasn’t removed during initial clay preparation. After sorption, the pH of the filtrates was always elevated relative to the introduced litter extract solution and did not vary across clay classes, averaging 6.51 ± 1.15 and 8.22 ± 0.28 for the eucalyptus and wheat filtrates, respectively (Fig. 4).

Solid-state 13C NMR spectra of filtrates from the four clay treatments at the mid-point of experiment. The initial litter extract DOC spectra are also given for reference

Solid-state 13C NMR spectra of the allophane (a), illite (b) and smectite (c) clay treatments before and after the experiment. No spectra were obtained for the oxide soil treatment due to large paramagnetic interferences

Molecular mixing model results for initial litter, week 5 extracts, week 5 filtrates and final soil samples for the a eucalyptus and b wheat treatments
Correlations with clay properties
Both the total amount of DOC retained and the percent of mineral retained DOC that was then respired during the course of the incubation (i.e. bioavailability) showed variable correlations with the measured clay properties with some differences between the two litter types (Table 2). Cation exchange capacity was not a good predictor of retention or bioavailability for either litter type. Retention of DOC was positively correlated with free sesquioxide concentration (Aldc and Fedc) and organo-metal complexes (Alpy) for wheat-derived DOC but non-significantly so for eucalyptus-derived DOC. Bioavailability of retained DOC originating from both litter types were strongly negatively correlated with Fepy and SRO mineral content (Al + Feox–py).
Discussion
Biochemistry of mineral-retained OM
The use of a 13C-enriched source for the DOC in this experiment has allowed the direct characterization of recently retained DOC onto aged mineral surfaces (as opposed to fresh minerals) via 13C NMR spectroscopy. The most striking feature of the spectra presented in Fig. 8 was in their similarity especially when compared to the disparate chemical composition of the litter sources (Fig. 3). This was readily apparent when a PCA was performed on the distribution of signal intensity across the eight major chemical shift regions (Fig. 10). In fact, a hierarchical cluster analysis found no significant differences between any of the soil samples (data not shown). In accord with previous research (Chorover and Amistadi 2001; Mikutta et al. 2007), there appears to be preferential retention of aromatic, phenolic and carboxylic-C during adsorption regardless of source DOC or clay type. This selective retention was most apparent in the results from the molecular mixing model analysis (Fig. 9) where a concentration of proteins and aliphatic C types was indicated in the soil core filtrates.

Results from a principal component analysis of the relative distribution of signal intensity across eight major chemical shift regions (eigenvectors shown as vectors on plot) for all samples
The chemical similarity of mineral-retained C regardless of source DOC chemistry or clay mineralogy suggests a commonality across purported binding mechanisms. In a study of DOC retention in tropical forest soils dominated by SRO minerals, mineral-retained C was also found to be dominated by partially oxidized aromatic acids with a high density of carboxylic functional groups (Kramer et al. 2012; Sanderman and Kramer 2013). In particular, a comparison of the PCA results presented in Fig. 10 in this study to an analysis presented by Kramer et al. (2012) is striking in similarity. These results are also consistent with the evidence presented by Keiluweit and Kleber (2009) that direct sorption of aromatic moieties to mineral surfaces due to aromatic pi-system electron donor–acceptor interactions are pervasive and important stabilization mechanisms operating in most soils.
In soils with a small fraction of highly reactive mineral phases, such as the illite and smectite treatments in this current study, the lower degree of observed stability (Fig. 6) may indicate that, if high DOC input rates are not sustained, over time the chemistry of mineral-retained OM will shift to an overall chemistry more similar to microbial biomass (i.e. alkyl and O-alkyl C dominated). This would be consistent with observations from fine fractions in several different soil orders (e.g. Rumpel et al. 2004; Schmidt et al. 2000; Schöning and Kögel-Knabner 2006).
Quantity and stability of mineral-retained OM
While similarly composed OM appears to be retained on all four clay types after our 11 week experiment (Figs. 8, 9, 10), the quantity and stability of the mineral-retained OM varied significantly (Fig. 6). Compared to the wheat-derived DOC, more eucalyptus-derived DOC was retained and, on average, a smaller proportion of this retained DOC was subsequently decomposed. We suggest here that these differences in the quantity and stability of mineral-retained OM are related to the both the quality of DOC produced from each litter source and the mechanisms controlling mineral-retention in the different clay mixtures.
Source litter controls
Microbial activity involved in both DOC production and in its initial rapid utilization will act to homogenize the biochemical composition of DOC as it enters the mineral soil relative to the chemical diversity found in the source materials (Sanderman and Kramer 2013). In this study, we found important contrasts between DOC produced from eucalyptus and wheat litters (Figs. 2, 3, 4). Despite similar quantities of DOC being produced from the two litters, the composition of DOC derived from the eucalyptus was concentrated in aromatic, phenolic and carboxylic C functional groups relative to the DOC extracted from wheat residues (Fig. 2) which was consistent with the much more acidic nature of the eucalyptus extracts (Fig. 4). These are the same functional groups that were selectively retained upon exposure to the clay minerals (Figs. 8, 10) and as a result, significantly more of the eucalyptus DOC was retained.
Interestingly, while the chemistry of the mineral retained DOC was basically indistinguishable between litter sources (Fig. 8), there were important differences in stability of the mineral retained DOC. A possible explanation for this difference is that the wheat litter produced a more degradable leachate higher in nutrients and lower in sparingly degradable substrates such as lignin-derived aromatic structures. As such, the microbial community living in the wheat DOC treatments may have been larger and more active than that found in the eucalyptus treatments. Then the combination of a more active microbial community with DOC retained by weaker retention mechanisms (see next section) can lead to the large differences in the decomposability of the mineral-retained OM that we observed for the wheat but not eucalyptus treatments.
Mineralogy controls
Results from this study indicate that the total amount of surface charge (represented by CEC) did not correlate with DOC retention dynamics rather it was the specific mineralogy that appeared to be the dominant driver of mineral retention and subsequent stability. Importantly, the correlation analysis (Table 2) revealed that organo-metal interactions and retention on SRO minerals were the properties that could explain most of the variability in retention and stability across the four clay types. In particular, the retained DOC was most stable when applied to minerals with an abundance of hydroxylated surfaces (the allophane and hydroxide treatments). Consistent with previous experimental studies (Gu et al. 1994; Mikutta et al. 2007), the surface chemistry of the different clay treatments suggested that retention of DOC due ligand exchange reactions was more stable that retention via cation/water bridging.
The reality of a complex heterogeneous physiochemical environment as found in soils is that multiple retention mechanisms will be operating simultaneously. In addition to the importance of ligand exchange reactions, aromatic cation-pi and pi–pi electron donor acceptor interactions are potential stabilizing mechanisms operating in these soils. Keiluweit and Kleber (2009) reported that bonding energies associated with aromatic pi-system interactions can be large enough to promote conformational changes which will render these aromatic moieties unavailable to enzymatic degradation.
Implications
There is an emerging view, primarily driven by compound-specific isotope tracer studies, that so-called stable carbon pools are microbial in origin and that there is rapid assimilation of plant-derived compounds into microbial communities (Gleixner 2013; Miltner et al. 2012). While these processes are undoubtedly important and may in fact be dominant in many soils, not all plant material is easily incorporated into microbial biomass. Results presented in this study and those of Kramer et al. (2012) suggest that in a humid leaching environment when reactive mineral phases are present not all plant-derived OM needs to pass through a microorganism before becoming stabilized. Rather, extracellular enzymatic activity, particularly peroxidise and ligninase, can act to mobilize and partially oxidize lignin polyphenols and other more recalcitrant plant-derived compounds (Schevchenko and Bailey 1996) and these highly reactive components of DOC can then bind directly to mineral surfaces, metals and existing organo-mineral complexes (Kaiser and Guggenberger 2000; Chorover and Amistadi 2001; Kramer et al. 2012). Once retained by minerals or metals, this DOC can be very stable and resistant to further degradation (Kramer et al. 2012; Sanderman and Amundson 2009). In this view of SOM dynamics, the microbial community is still the major driver of OM cycling but we recognize that a variable but significant fraction of plant-derived OM does not have to be incorporated into microbial biomass before becoming stabilized in soils, especially those soils containing highly reactive mineral phases.
References
Arnarson TS, Keil RG (2000) Mechanisms of pore water organic matter adsorption to montmorillonite. Mar Chem 71:309–320
Baldock JA, Skjemstad JO (2000) Role of the soil matrix and minerals in protecting natural organic materials against biological attack. Org Geochem 31:697–710
Baldock JA, Masiello CA, Gélinas Y, Hedges JI (2004) Cycling and composition of organic matter in terrestrial and marine ecosystems. Mar Chem 92:39–64
Baldock JA, Sanderman J, Macdonald LM, Puccini A, Hawke B, Szarvas S, McGowan J (2013) Quantifying the allocation of soil organic carbon to biologically significant fractions. Soil Res 51:561–576
Butler JHA, Eickhoff CP (1979) A simple cabinet for growing C-14 labelled plant material. Lab Pract 28:28–30
Chapman HD (1965) Cation-exchange capacity. In: Black CA, Evans DD, White JL, Ensminger LE, Clark FE (eds) Methods of soil analysis. Part 2. American Society of Agronomy, Madison, pp 899–904
Chorover J, Amistadi MK (2001) Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces. Geochim Cosmochim Acta 65:95–109
Chorover J, Amistadi MK, Chadwick OA (2004) Surface charge evolution of mineral-organic complexes during pedogenesis in Hawaiian basalt. Geochim Cosmochim Acta 68:4859–4876
Clark KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143
Coplen TB (2011) Guidelines and recommended terms for expression of stable-isotope-ratio and gas-ratio measurement results. Rapid Commun Mass Spectrom 25:2538–2560
De Troyer I, Bouillon S, Barker S, Perry C, Coorevits K, Merckx R (2010) Stable isotope analysis of dissolved organic carbon in soil solutions using a catalytic combustion total organic carbon analyzer-isotope ratio mass spectrometer with a cryofocusing interface. Rapid Commun Mass Spectrom 24:365–374
Eusterhues K, Rumpel C, Kleber M, Kögel-Knabner I (2003) Stabilisation of soil organic matter by interactions with minerals as revealed by mineral dissolution and oxidative degradation. Org Geochem 34:1591–1600
Feng XJ, Simpson AJ, Simpson MJ (2005) Chemical and mineralogical controls on humic acid sorption to clay mineral surfaces. Org Geochem 36:1553–1566
Gleixner G (2013) Soil organic matter dynamics: a biological perspective derived from the use of compound-specific isotopes studies. Ecol Res 28:683–695
Gu BH, Schmitt J, Chen ZH, Liang LY, McCarthy JF (1994) Adsorption and desorption of natural organic-matter on iron-oxide: mechanisms and models. Environ Sci Technol 28:38–46
Kaiser K, Guggenberger G (2000) The role of DOM sorption to mineral surfaces in the preservation of organic matter in soils. Org Geochem 31:711–725
Kaiser K, Kalbitz K (2012) Cycling downwards: dissolved organic matter in soils. Soil Biol Biochem 52:29–32
Kaiser K, Guggenberger G, Haumaier L, Zech W (2002) The composition of dissolved organic matter in forest soil solutions: changes induced by seasons and passage through the mineral soil. Org Geochem 33:307–318
Kalbitz K, Kaiser K (2008) Contribution of dissolved organic matter to carbon storage in forest mineral soils. J Plant Nutr Soil Sci (Zeitschrift Fur Pflanzenernahrung Und Bodenkunde) 171:52–60
Keiluweit M, Kleber M (2009) Molecular-level interactions in soils and sediments: the role of aromatic pi-systems. Environ Sci Technol 43:3421–3429
Kindler R, Siemens J, Kaiser K, Walmsley DC, Bernhofer C, Buchmann N, Cellier P, Eugster W, Gleixner G, Grũnwald T, Heim A, Ibrom A, Jones SK, Jones M, Klumpp K, Kutsch W, Larsen KS, Lehuger S, Loubet B, McKenzie R, Moors E, Osborne B, Pilegaard K, Rebmann C, Saunders M, Schmidt MWI, Schrumpf M, Seyfferth J, Skiba U, Soussana JF, Sutton MA, Tefs C, Vowinckel B, Zeeman MJ, Kaupenjohann M (2011) Dissolved carbon leaching from soil is a crucial component of the net ecosystem carbon balance. Glob Change Biol 17:1167–1185
Kleber M, Sollins P, Sutton R (2007) A conceptual model of organo-mineral interactions in soils: self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 85:9–24
Kögel-Knabner I, Ekschmitt K, Flessa H, Guggenberger G, Matzner E, Marschner B, von Luetzow M (2008) An integrative approach of organic matter stabilization in temperate soils: linking chemistry, physics, and biology. J Plant Nutr Soil Sci (Zeitschrift Fur Pflanzenernahrung Und Bodenkunde) 171:5–13
Kramer MG, Sanderman J, Chadwick OA, Chorover J, Vitousek PM (2012) Long-term carbon storage through retention of dissolved aromatic acids by reactive particles in soil. Glob Change Biol 18:2594–2605
Leifeld J, Kögel-Knabner I (2001) Organic carbon and nitrogen in fine soil fractions after treatment with hydrogen peroxide. Soil Biol Biochem 33:2155–2158
Michalzik B, Tipping E, Mulder J, Lancho JFG, Matzner E, Bryant CL, Clarke N, Lofts S, Esteban MAV (2003) Modelling the production and transport of dissolved organic carbon in forest soils. Biogeochemistry 66:241–264
Mikutta R, Kleber M, Kaiser K, Jahn R (2005) Review: organic matter removal from soils using hydrogen peroxide, sodium hypochlorite, and disodium peroxodisulfate. Soil Sci Soc Am J 69:120–135
Mikutta R, Mikutta C, Kalbitz K, Scheel T, Kaiser K, Jahn R (2007) Biodegradation of forest floor organic matter bound to minerals via different binding mechanisms. Geochim Cosmochim Acta 71:2569–2590
Mikutta R, Schaumann GE, Gildemeister D, Bonneville S, Kramer MG, Chorover J, Chadwick OA, Guggenberger G (2009) Biogeochemistry of mineral-organic associations across a long-term mineralogical soil gradient (0.3–4,100 kyr), Hawaiian Islands. Geochim Cosmochim Acta 73:2034–2060
Miltner A, Bombach P, Schmidt-Brucken B, Kastner M (2012) SOM genesis: microbial biomass as a significant source. Biogeochemistry 111:41–55
Neff JC, Asner GP (2001) Dissolved organic carbon in terrestrial ecosystems: synthesis and a model. Ecosystems 4:29–48
Piccolo A (2001) The supramolecular structure of humic substances. Soil Sci 166:810–832
Qualls RG (2000) Comparison of the behavior of soluble organic and inorganic nutrients in forest soils. For Ecol Manag 138:29–50
Römkens P, Dolfing J (1998) Effect of Ca on the solubility and molecular size distribution of DOC and Cu binding in soil solution samples. Environ Sci Technol 32:363–369
Rumpel C, Eusterhues K, Kögel-Knabner I (2004) Location and chemical composition of stabilized organic carbon in topsoil and subsoil horizons of two acid forest soils. Soil Biol Biochem 36:177–190
Sanderman J, Amundson R (2009) A comparative study of dissolved organic carbon transport and stabilization in California forest and grassland soils. Biogeochemistry 92:41–59
Sanderman J, Kramer MG (2013) Differential production yet chemical similarity of dissolved organic matter across a chronosequence with contrasting nutrient availability in Hawaii. Biogeochemistry 113:259–269
Sanderman J, Baldock JA, Amundson R (2008) Dissolved organic carbon chemistry and dynamics in contrasting forest and grassland soils. Biogeochemistry 89:181–198
Scheel T, Dorfler C, Kalbitz K (2007) Precipitation of dissolved organic matter by aluminum stabilizes carbon in acidic forest soils. Soil Sci Soc Am J 71:64–74
Scheel T, Haumaier L, Ellerbrock RH, Ruhlmann J, Kalbitz K (2008) Properties of organic matter precipitated from acidic forest soil solutions. Org Geochem 39:1439–1453
Schmidt MWI, Knicker H, Kögel-Knabner I (2000) Organic matter accumulating in Aeh and Bh horizons of a Podzol: chemical characterization in primary organo-mineral associations. Org Geochem 31:727–734
Schmidt MWI, Torn MS, Abiven S, Dittmar T, Guggenberger G, Janssens IA, Kleber M, Kögel-Knabner I, Lehmann J, Manning DAC, Nannipieri P, Rasse DP, Weiner S, Trumbore SE (2011) Persistence of soil organic matter as an ecosystem property. Nature 478:49–56
Schnitzer M (2000) A lifetime perspective on the chemistry of soil organic matter. Adv Agron 68(68):1–58
Schöning I, Kögel-Knabner I (2006) Chemical composition of young and old carbon pools throughout Cambisol and Luvisol profiles under forests. Soil Biol Biochem 38:2411–2424
Sheldrick BH (1984) Acid ammonium oxalate-extractable Fe and Al (Mn and Si if desired). LRRI: 84-30. Land Resource Research Institute, Ottawa
Shevchenko SM, Bailey GW (1996) Life after death: lignin-humic relationships reexamined. Crit Rev Environ Sci Technol 26:95–153
Smernik R, Skjemstad J (2009) Mechanisms of organic matter stabilization and destabilization in soils and sediments: conference introduction. Biogeochemistry 92:3–8
Stevenson FJ (1982) Humus chemistry: genesis, composition, reactions. Wiley, New York
Sutton R, Sposito G (2005) Molecular structure in soil humic substances: the new view. Environ Sci Technol 39:9009–9015
Sutton R, Sposito G (2006) Molecular simulation of humic substance-Ca-montmorillonite complexes. Geochim Cosmochim Acta 70:3566–3581
Theng BKG, Churchman GJ, Newman RH (1986) The occurrence of interlayer clay-organic complexes in 2 New Zealand soils. Soil Sci 142:262–266
Tucker ME (1988) Techniques in sedimentology. Blackwell Scientific Publications, Oxford, p 394
Weishaar JL, Aiken GR, Bergamaschi BA, Fram MS, Fujii R, Mopper K (2003) Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon. Environ Sci Technol 37:4702–4708
Wershaw RL, Llaguno EC, Leenheer JA (1996) Mechanism of formation of humus coatings on mineral surfaces. 3. Composition of adsorbed organic acids from compost leachate on alumina by solid-state (13)C NMR. Colloids Surf A 108:213–223
Acknowledgments
This work was funded by a strategic appropriation grant from the CSIRO Sustainable Agriculture National Research Flagship. We would like to thank M Raven for XRD analysis, C Creamer, L Macdonald and M Farrell for thoughtful discussions during the development of this manuscript and an anonymous reviewer for insightful comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Jan Mulder
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sanderman, J., Maddern, T. & Baldock, J. Similar composition but differential stability of mineral retained organic matter across four classes of clay minerals. Biogeochemistry 121, 409–424 (2014). https://doi.org/10.1007/s10533-014-0009-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-014-0009-8