
Abstract
Field-scale observations of two upland soils derived from contrasting granite and basalt bedrocks are presented to hypothesize that redox activity of rhizospheres exerts substantial effects on mineral dissolution and colloidal translocation in many upland soils. Rhizospheres are redox-active microsites and in the absence of O2, oxidation of rhizodeposits can be coupled by reduction of redox-active species such as Fe, a biogenic reduction that leads to Fe translocation and oxidation, accompanied by substantial proton flux. Not only do rhizogenic Fe–C redox cycles demonstrate a process by which the rhizosphere affects an environment well outside the near-root zone, but these redox processes are also hypothesized to be potent weathering systems, such that rhizogenic redox-reactions complement acid- and ligand-promoted reactions as major biogeochemical processes that control crustal weathering. The potential significance of Fe–C redox cycling is underscored by the deep and extensive rooting and mottling of upland subsoils across a wide range of plant communities, lithologies, and soil-moisture and temperature regimes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organic-carbon mediated mineral weathering in soils and sediments are long studied, and the predominant processes driving the chemical weathering of the Earth’s crust are considered to be acid- and ligand-promoted dissolution reactions (Qualls and Haines 1991; Stone 1997; Raulund-Rasmussen et al. 1998; Richter and Markewitz 2001). Here, we hypothesize that rhizosphere-derived organic reductants in concert with dissimilatory Fe-reducing microorganisms set in motion a series of physical and chemical reactions in which rhizospheres become Fe-depleted and carbon- and clay-enriched, and environments removed from roots become Fe-enriched and mineral- and clay-depleted. These rhizogenically initiated reactions represent a biogeochemical mechanism by which weathering reactions are directly affected outside the rhizosphere, hypothetically a mechanism by which soil mineralogy is altered and re-structured throughout the entire soil volume, in the rhizosphere and the surrounding soil volumes alike.
Organic reductants are ubiquitous, although unevenly distributed in soils and sediments. They participate in redox reactions in a number of elemental cycles, including those of O2, Fe, N, S, Mn, Zn, Cu, Cr, and U (Schulten and Schnitzer 1998; Buerge and Hug 1998; Liger et al. 1999; Hagedorn et al. 2000; Michalzik et al. 2001; Roden et al. 2004; Chacon et al. 2006; Thompson et al. 2006a, b). In the natural environment, organic matter serves as an electron donor upon oxidative decomposition. But only recently has organic matter been appreciated to be an electron shuttle which, via microbially driven transformations, may donate and accept electrons, thereby reducing and oxidizing redox-active metals (Lovley et al. 1996, 1998; Scott et al. 1998; Fimmen et al. 2007). In this paper, we focus on C and the redox-active metal Fe, due to iron’s environmental ubiquity and acknowledge that other redox-active elements are also relevant to soil redox processes.
The relationship between soil organic matter (OM), O2, Fe, pH, and redox chemistry have long been studied (Schlichting and Schwertmann 1973; Emerson et al. 1999; Vepraskas 2001; Neubauer et al. 2002; Roden et al. 2004, Kirk 2004), with attention focused on hydromorphic soils and sediments (Kirk 2004). Under anoxic conditions, plants often transport atmospheric O2 to their roots to maintain aerobic respiration, and rhizospheres become microsites inhabited by aerobic microbes that oxidize aqueous ferrous Fe and precipitate Fe oxides, resulting in the so-called Fe plaque and features such as “pipestems” (Bidwell et al. 1968; Emerson et al. 1999; Maurice et al. 2000; Maurice 2002; Neubauer et al. 2002; Glasauer et al. 2003; Weiss et al. 2003, 2004).
Here, we propose to expand the concept of rhizogenic redox reactions to be significant biogeochemical reactions not only in aquic and poorly drained ecosystems, but in many imperfectly and even well drained upland ecosystems as well. Even well drained upland soils have subsoil environments in which O2 and its diffusion from the aboveground atmosphere are periodically unable to keep pace with the demands of the root and decomposer systems to accept electrons (Silver et al. 1999). As a consequence, we hypothesize that in a wide range of upland ecosystems, oxidation of rhizogenically derived organic reductants periodically drives reduction of Fe with great consequences on the environment. Although root-affected reduced microsites have been described in some detail (Vepraskas 2001), the redoximorphic features proposed here are believed to originate from the oxidation of organic matter derived from living and dead root systems, and to set off a sequence of reactions that leads to Fe oxidation away from the root, substantial proton flux, and translocation of solutes and colloids both away from and into the rhizosphere itself. In addition, we show that morphologically, rhizospheres in upland subsoils are carbon- and clay-enriched and Fe-depleted, and inter-rhizosphere microsites remain carbon-poor but become Fe-enriched and clay-depleted.
The proposed biogeochemical weathering mechanism may well have positive feedbacks, which promote conditions for ongoing Fe reduction and oxidation, all assuming a continued supply of organic reductants. These processes have the potential to affect the dissolution and precipitation of Fe oxides, alumino-silicate clays, and a variety of primary minerals, as well as the sequestration of organic carbon deep within the mineral soil. This model is consistent with laboratory-scale observations of Fe redox behavior (Thompson et al. 2006a, b), and bears relation to pedological concepts of Fe-mediated mineral weathering in rice paddies that undergo seasonal flooding and drainage (Brinkman 1970; van Breeman 1988).
Materials and methods
Site descriptions, and sample acquisition and preparation
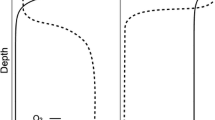
In two upland forest ecosystems, soils were deeply excavated with a backhoe (>2-m); in a granite-gneiss-derived Ultisol in the Calhoun Experimental Forest, Union County, South Carolina, USA, and in a basalt-derived Alfisol in the Duke Forest, Durham County, North Carolina, USA. Both soils are residual, derived directly from bedrock below, and have well developed horizonation (Figs. 1 and 2). Both soils are ancient, forming in mainly warm humid climates under forest vegetation, on broad geomorphically stable interfluves with <5% slope, and with permanent groundwater >5 m depth under current climates. Despite the great age of these soils, bedrock effects remain strong: the granitic gneiss under the Calhoun soil and the diorite under Duke soil have left legacies on both structure and process of these soil environments.

(a) Soil profile (0 to >240-cm) at the Calhoun term soil experiment. The loblolly pine forest (about 45 years in age) is supported by a soil that was cultivated for cotton continuously since the 1930s and periodically since 1810 (Richter and Markewitz 2001). The deep, acidic Ultisol is derived from granitic-gneiss bedrock, and has depth-dependent horizons that contrast in texture, mineralogy, structure, and biogeochemical processes. (b) The striking color-contrasts of rhizospheres and inter-rhizosphere microsites suggest a Liesegang-banding phenomena (Chan et al. 2007)

(a) Soil profile (0 to >96 cm) at the Duke Forest. The mixed hardwood forest, primarily white oak (Quercus alba L.) is supported by a soil that has been grazed, used for a woodlot but probably not extensively cultivated. The deep, non-acidic Alfisol is derived from basalt bedrock, and has depth-dependent horizons that contrast in texture, mineralogy, structure, and biogeochemical processes. (b) Samples of microsites in B horizons (rhizospheres and Fe concentrations) were carefully collected with knives and scalpels. Though the morphological patterns of the Duke Forest soil contrasts with those of the Calhoun, the geochemistry of subsoil rhizospheres and Fe-rich microsites is strikingly similar to those in Fig. 1 (Tables 2 and 3)
Vegetation growing on the Calhoun soil is secondary loblolly pine (Pinus taeda L.), planted in 1957 following long cultivation mainly for cotton. Soils supporting this pine forest are classified as the Appling series, Typic Kanhapludults. Relatively coarse-textured and porous A and E horizons overlie acidic, kandic Bt-horizons that are dominated by kaolinite and Fe and Al oxides. Vegetation at the Duke Forest site is mixed hardwood, primarily white oak (Quercus alba L.), supported by Enon series soils that are classified as Ultic Hapludalfs with near-neutral pH regimes, and medium-textured and porous well structured A and AB horizons that overlie smectitic Bt horizons that like the Calhoun soil have prominent Fe and Al oxides.
Both soils are well described in physical and chemical properties and their dynamics of biogeochemical change on time scales of years and decades (Markewitz et al. 1998; Markewitz and Richter 2000; Richter and Markewitz 2001; Richter et al. 2007; Li et al. 2008; Oh and Richter, 2005; Oren et al. 2001; Schlesinger et al. 1999). All previous studies, however, have examined bulk soil chemistry of composited samples of whole soil horizons and soil layers. None of our previous studies have sampled the heterogeneity of microsites within the soil environment, as we do in this paper.
Soils at both sites were sampled from whole genetic horizons and from rhizosphere-associated microsites (both rhizospheres themselves and surrounding soil volumes, details given below). At the Calhoun site, depth-integrated, whole-horizon samples were taken from the O, A (0–0.1 m), E (0.1–0.4 m), EB (0.4–0.8 m), Bt (0.8–1.5 m), BC (1.5–2.0 m), and CB (2.0–2.5 m) horizons (Table 1). At the Duke Forest site, depth-integrated, whole-horizon samples were taken from the A (0–0.2 m), BA (0.2–0.5 m), B (0.5–0.9 m), Bt (0.9–1.3 m), and Bt (1.3–1.5 m) horizons. In both soils, scalpels and knives were used to carefully collect the two distinctively contrasting microsites within Bt horizons, sampling rhizospheres and surrounding volumes of oxidized material (Figs. 1 and 2). Fine rootlets and fragments of organic matter were removed from the rhizosphere samples. Depth-integrated and microsite samples were collected from Bt horizons at 0.8–1.5 m at the Calhoun Forest (Table 2) and 0.9–1.5 m at the Duke Forest (Table 3). Fe-depleted low-chroma rhizospheres from the Bt-horizons had Munsell colors when field moist to be 5Y 8/2 and 10 YR 8/1 or “light gray” for the Calhoun and Duke Forest soils. The oxidized microsites (referred to as “redox concentrations” or “Fe concentrations” according to Vepraskas (1992, 2001)) were 2.5YR 5/8 and 10YR 6/8 or orange and bright yellowish brown for the two soils, respectively. Due to these distinct color differences between the rhizosphere and their surrounding oxidized material, we henceforth refer to the samples from the rhizosphere (intra-rhizosphere environments) as “rhizosphere” or “gray” microsites and the microsites surrounding the rhizosphere (inter-rhizosphere or non-rhizosphere environments) as “Fe oxide-rich” or “orange” microsites.
Soil horizon samples were air-dried and seived with a 2-mm screen prior to analyses. Microsite samples, however, were subdivided upon return to the lab, with one part air dried and sieved with a 2-mm screen, prior to estimating texture, whole soil C and N, 14C, extractable Fe and Al oxides, effective cation exchange capacity (ECEC), pH, exchangeable acidity and base cations, and mineralogy. A second part of the microsite samples was kept field moist, though stored on dry ice and shipped overnight for fatty acid methyl ester (FAME) analyses which took place 48 h after receipt during which time the samples were stored at −80°C. A third part of the microsite samples was not screened and was kept field fresh and examined within a week of collection by environmental scanning electron microscopy (ESEM).
Soil chemical and physical properties
Texture was analyzed by pipette methods following mechanical and chemical dispersion using sodium hexametaphosphate (Page 1982). Soil pH (pHs) was measured in duplicate with an Orion 320 pH-meter fitted with a VWR Scientific pH electrode, by suspending samples in 0.01 M CaCl2 (1:10 m/v soil:solution ratio). Concentrations of total C and N were measured in triplicate with a CE Instruments Flash EA 1112 series CHN analyzer using acetanilide as a check standard.
Iron and Al oxides were characterized using two extractions to evaluate both short-range order, SRO, and more crystalline oxides. The SRO Fe and Al oxides were extracted using acidified ammonium oxalate (AAO) adjusted to pH 3 as outlined in Loeppert and Inskeep (1996). Soil samples were extracted with AAO solution for 2 hours in the dark, with suspensions centrifuged and filtered. Filtrates were analyzed for Fe and Al by atomic absorption spectroscopy (AAS 5100PC, Perkin Elmer, Norwalk, Connecticut). More crystalline Fe and Al oxides were extracted from the Calhoun soils with dithionite-citrate-bicarbonate (DCB) solutions (Mehra and Jackson 1960; Loeppert and Inskeep 1996) and from the Duke Forest soils with dithionite-citrate (DC) solutions (Page 1982). Both DCB and DC extractions subject SRO and more crystalline forms of soil Fe oxides to reductive dissolution. Briefly, DCB extractions samples were heated to 75–80°C in the citrate-bicarbonate buffer, and suspensions reduced by repeatedly adding 1 g of sodium dithionite (Na2S2O4) every 15 min up to four occasions. For DC extractions, methods were similar to those of DCB, although without the bicarbonate buffer which is considered unnecessary. Suspensions were centrifuged, filtered, and analyzed by AAS for Fe and Al. Extractable oxide phases are referred to as Feo and Alo for oxalate extractable fractions and Fed and Ald for dithionite extractable fractions.
Soil exchangeable acidity, aluminum acidity, and total acidity were determined by extraction and titration methods. KCl-exchangeable acidity was extracted with 1 M KCl in a 1:10 (m/v) soil to solution ratio (Thomas 1982) followed by titration with 0.02 M sodium hydroxide to an endpoint of 8.2. Exchangeable Al was subsequently estimated in the neutralized KCl extract by addition of 1 M KF to each solution and back titration with 0.02 M HCl to an endpoint of pH 8.2. Total acidity was extracted with a BaCl2 that was adjusted to pH 8.2 and buffered with triethanolamine (Thomas 1982), and the filtrate titrated with 0.01 M HCl to an endpoint of pH 5.0.
Exchangeable cations were extracted with 1 M ammonium acetate (NH4OAc) adjusted to pH 7 in a 1:20 (m/v) soil to solution ratio, which was shaken for 30min, centrifuged at 3400 rpm for 10 min and gravity filtered. Solution cation concentrations (Ca, Mg, and K) were determined by atomic absorption spectroscopy. Effective and total cation exchange capacity and base saturation were estimated from NH4OAc extractable cations summed with KCl exchangeable acidity or BaCl2–TEA acidity, respectively.
Soil mineralogy by X-ray diffraction and differential scanning calorimetry
Samples from the Calhoun soil were analyzed for mineralogy by X-ray diffraction (XRD) using a Philips PW 1729 X-ray generator (CuKα radiation) with a curved crystal monochrometer set at 35 kV and 20 mA. Samples were analyzed by firmly pressing pulverized material in powder-mount slides with both analyzed from 8° to 80° 2θ at a rate of 0.020° 2θ per second. Samples were analyzed for Fe and Al oxides by selective reductive-dissolution with DCB and selective dissolution with NaOH, respectively (Shaw 2001). The clay fraction (<2 μm) was isolated from gray microsites by repeated suspension and centrifugation, and analyzed for the presence of hydroxy-interlayered vermiculite (HIV) and gibbsite from 3° to 32° 2θ at reduced speed (0.15° 2θ per second). Sample treatments are as follows: Mg-saturation and ethylene glycol solvation (MgEg) to expand collapsible minerals; K-saturation at room temperature (K-25) to collapse expandable minerals; K-saturation heated to 100°C (K-100) to collapse hydroxy-Al interlayered minerals; K-saturation heated to 300°C (K-300) to decompose gibbsite; and K-saturation heated to 550°C (K-550) to decompose kaolinite. A quartz standard was analyzed from 8° to 80° 2θ at 0.020° 2θ per second. Material isolated from both microsites were further investigated for gibbsite, HIV, and kaolinite by differential scanning calorimetery (DSC) using a Thermal Instruments DSC model 2920, with flowing N2(g) at 448.2 kPa (65 psi).
Radiocarbon analysis
Carbon isotopic ratios were determined at the University of Georgia Accelerator Mass Spectrometry facility (Athens, GA) using a National Electrostatics Corporation Model 1.5SDH-1 Pelletron Accelerator Mass Spectrometer. Samples were air-dried, sieved for fine roots, and delivered to the University of Georgia’s accelerator mass spectrometry (AMS) facility for analysis without further treatment. Graphite targets were prepared from soil organic matter and data are reported as Δ14C, the per mil deviation of 14C/12C compared with a decay-corrected oxalic acid standard. Negative values indicate predominance of old soil organic matter with 14C that has experienced significant radioactive decay (half-life of 5,730 years), whereas positive values of Δ14C indicate presence of thermonuclear bomb-produced 14C (Trumbore 1996). Due to the acidity of these soils (Table 2, Richter and Markewitz 1995), soil carbon can be assumed to be organic and devoid of carbonates.
Environmental scanning electron microscopy
Environmental scanning electron microscopy (ESEM) was used to qualitatively characterize contrasting surface topography of both microsites. The ESEM examined fully hydrated samples which were sampled directly onto dry ice in an attempt to preserve the native soil structure and aggregates. As such, distortion or destruction of samples, resulting from standard preparation techniques required for SEM, was avoided. ESEM spectra were obtained using a Philips XL 30 ESEM TMP at a pressure of 66.7 Pa (0.5 torr). Images were analyzed using AnalySiS image analysis and editing software package.
Fatty acid methyl-ester analysis
Fatty acid methyl-ester (FAME) extractions were conducted according to the method utilized by Schutter and Dick (2002). All solvents and chemicals used were of analytical grade, and glassware was cleaned using a 3-stage system, first with detergent, followed by methanol and acetone, with each stage rinsed with Millipure H2O. One gram of soil was placed in a 40 ml scintillation vial and submersed in 5 ml of 0.2 M KOH dissolved in methanol and sonicated for 1 h. About 1 ml of 1 M acetic acid was added to neutralize KOH and samples were mixed using a vortex for 1 min. A 3.3 ml aliquot of hexane was added and mixed on a vortex for 1 min, followed by 1.67-ml of Millipure water to separate phases. The samples were mixed a third time for 1 min on a vortex and then centrifuged at 2500 rpm for 15 min. The hexane layer (5 ml) was removed using a pipette and placed in a 7 ml disposable glass tube. Samples were completely dried under a gentle stream of 99.999% N2(g), and the residue re-dissolved in 150 μl of hexane and placed in a 200 μl glass insert and fitted into 2 ml GC vial for analysis.
Samples were analyzed using a Polaris Q Ion Trap mass spectrometer-gas chromatograph (Thermo Electron Corp). An HP-5 MS 5%-phenyl-methylpolysiloxane (25 m) capillary column (Agilent Inc, Palo Alto, CA) was used to analyze 1 μl samples in splitless mode at a constant He flow of 1.1 ml min−1 and initial temperature of 120°C for 0.5 min which was ramped from 120 to 260°C at a rate of 5°C min−1 with a 5 min hold at 280°C. Standard mixtures containing 37 and 24 fatty acid methyl esters were used to confirm peak identification (Supelco, Inc., Bellefonte, PA). Fatty acid methyl esters as well as other organic fragments recovered during the alkaline-methanol hydrolysis were further confirmed by using the NIST mass spectral database (Thermo Electron Corp.), and other identification resources useful for identifying fatty acid methyl ester fragment patterns (Murphy 1993) in mass spectra. Similar to the use of phospholipid fatty acids (PLFAs) which are considered an indicator of living microbial community biomass, FAME’s are also considered an acceptable, representative, and comparable method for assessing the microbial community members in soils, particularly for soils with low microbial biomass (Dierksen et al. 2002; Schutter and Dick 2002).
Results
Observations of profiles, horizons, and microsites
In both Calhoun and Duke soils, the forest ecosystems’ surficial O and A horizons are intensively rooted and are the most active volumes of organic matter deposition and decomposition (Table 1, Figs. 1 and 2). These layers have prominent permeability and aeration not only due to intensive tree rooting but also due to the Calhoun’s sandy loam texture and the Duke soil’s well developed structure. Underlying B horizons of both soils are clay-rich and Fe- and Al-oxide coated, though with contrasting clay mineralogy, kaolinite-dominated in Calhoun soils and smectite-dominated in the Duke Forest (Table 1). Both soils have udic moisture regimes, and are generally well oxygenated, although both periodically perch precipitation water within B and BC horizons and during these periods gas diffusivity slows considerably (Oh and Richter 2005; Richter et al. 2007). Therefore, the profiles’ minimum hydraulic conductivity values are likely in the lower B and BC horizons (O’Brien and Buol 1984; West et al. 2008).
While organic matter, and root biomass and activity are concentrated in surficial horizons, organic reductants are deposited at depth, especially by deep tree roots that extend into and through underlying B horizons, which in both soils is to >2-m in depth (Richter and Markewitz 2001; Oh and Richter 2005). Tree roots access nutrients and water from subsoils, and are particularly important late in the growing season when more surficial layers tend to be exhausted of bio-available water. In subsoils, roots and rhizospheres are not only sources of organic reductants but also respiration hotspots and O2 sinks.
Redoximorphic features provide visual evidence for electron acceptors shifting from O2 to Fe within B horizons of both soils (Figs. 1 and 2). Calhoun and Duke subsoil rhizospheres have Munsell chromas of 1 or 2 and values of 8, and are surrounded by Fe concentrations (Vepraskas 2001) that are chroma 8 and oxidized in appearance. When samples of the Calhoun microsites are observed with ESEM, prolific rooting and hyphal networks of mycorrhizal fungi are obvious throughout the gray rhizospheres (Fig. 3a, b), observations in stark contrast to the absence of roots and hyphae in immediately adjacent Fe oxide-rich samples (Fig. 3c, d). The fungal hyphae were morphologically identified in ESEM images as Rhizopogon spp (Prof. Rytas Vilgalys, pers. commun, Durham, NC), a pine mycorrhizal fungi. Fungi were also identified by an 18:2 fatty acid biomarker in FAME analysis with GC-MS detection (Fig. 4). In fact, the fatty acids extractable from the grey microsites with alkaline methanol were overwhelmingly mono-unsaturated, and cyclopropyl, consistent with fatty acids that are sometimes associated with bacteria that have the capacity to reduce Fe(III), e.g. Geobacter (Lovley et al. 1993; Zhang et al. 2003). Remarkably, these contrasting microsite environments, i.e. subsoil rhizospheres and surrounding Fe oxide rich (non-rhizosphere) environments, are immediately adjacent, within millimeters, and are microhabitats separated by what must be extremely steep biogeochemical gradients (Figs. 1 and 2).

Environmental scanning electron microscopic (ESEM) images of gray (panels a and b) and orange (panels c and d) microsites. Abundant ectomycorrhizal hyphae permeate pores of the gray microsite (a, b) and apparent bacterial cocci are seen in the orange microsite (d). Scale bars represent 100, 20, 10, and 2 μm for panels a, b, c, and d, respectively

Gas chromatogram of fatty acid methyl ester profile (FAME) isolated from both soil microsites
In both Calhoun and Duke Forest soils, organic carbon is 2- to 3-fold higher in rhizosphere samples compared with those taken from the surrounding Fe oxide-rich microsites (Tables 2 and 3), a difference attributed to rhizodeposition. Radiocarbon analysis of organic matter in Calhoun Bt horizons (0.8 to 1.5-m depth) indicates dramatic differences in carbon ages between the two microsites, and therefore an apparent lack of carbon mixing (Table 2). Gray rhizospheres in the Calhoun soil had a much more modern organic C than that in orange microsites: Δ14C values of −658‰ vs. −244‰, respectively (Table 2). The relatively more modern signature of the C in rhizosphere organic matter indicates sequestration of bomb−14C by the roots of the contemporary Calhoun pine ecosystem, a pine forest planted in 1957 in long-cultivated cotton fields (Richter et al. 1999). The 50-year old pine forests can be safely assumed to root much more deeply than cotton whose roots are highly sensitive to subsoil acidity (Foy 1978), and in fact the radiocarbon and total C data suggest that the pine forest has sequestered considerable C in deep subsoil rhizospheres (Richter et al. 1999). Assuming a bi-modal distribution of 14C-ages within the gray microsites, with the old component being the ancient carbon within the orange microsites (0.081% C, Δ14C of −657.8‰), the additional carbon within the gray microsites (0.12%) is estimated to have a Δ14C of +38.5‰, and is therefore taken to be C added by the contemporary pine rhizospheres of the last 50 years.
The quality of subsoil organic matter was also informative in its concentrations of fatty acids associated with fungi and Fe-reducing bacteria. Alkaline methanol extractions in the FAME analysis yielded quinoid fragments, specifically 2,5-dialkyl substituted 1,4-benzoquinone derivatives, as identified through the NIST database by MS fragmentation patterns. These observations suggest that phenolic moieties (reduced quinones or hydroquinones) may participate as part of the bioorganic reduction that transfers electrons to electron-deficient metals. The observation of quinoid compounds in terrestrial organic matter is consistent with results of recent studies indicating that quinone–hydroquinone redox couples are prominent redox-active components in natural organic matter (Lovley et al. 1996; Scott et al. 1998; Fimmen et al. 2007). More research on electron budgeting and the role of quinone-phenolic moities in electron transfer processes will greatly increase our understanding of soils as decomposer systems.
The striking color contrasts between the rhizosphere and non-rhizosphere microsites indicate substantial translocations of Fe from rhizospheres to soil volumes outside the rhizosphere are substantial in both Calhoun and Duke Forest subsoils (Figs. 1 and 2; Tables 2 and 3). In both upland soils, extractable Fe in rhizospheres was only about 15% of that in the surrounding soil volumes of Fe concentrations. In the Duke Forest soil, extractable Mn in rhizosphere microsites was only about 5% of that in surrounding Fe oxide-rich microsites (Table 3). Oxidized forms of Fe and Mn function as terminal electron acceptors in microbial respiration, and we attribute their depletion within the rhizosphere to reductive dissolution rather than organo-complexation. The distinctly different 14C ages of soil C in the two microsites argues against chelation-assisted Fe(III)-oxide dissolution and translocation as the major mechanism explaining these C and Fe profiles. Complexation-facilitated dissolution would likely produce less distinct contrasts in Δ14C and soil organic C concentrations in the two microsites.
As striking as the contrasts of color, C, biota, and Fe oxides, are the textural contrasts between rhizosphere and Fe oxide-rich microsites. Soil within rhizospheres is two- to three-fold higher in clay compared with Fe oxide-rich microsites: 73.0 vs 26.6% clay in Calhoun soils and 59.6 vs 30% clay in Duke Forest samples, respectively (Tables 2 and 3). The relatively coarser textures of Fe oxide-rich microsites appear not to be the consequence of Fe oxide precipitations consolidating clay particles, as samples of Fe-rich microsites stripped of Fe by dithionite reduction increased the clay fraction by only a few percent (Table 4).
In the Calhoun rhizospheres, clay minerals which are mainly kaolinite appear well-oriented (Fig. 3a, b). In orange microsites, pitted and etched features characterize mineral Fe oxide’s precipitation (Fig. 3c, d), and goethite and hematite are readily identified by XRD (Fig. 5). An absence of sharp well-defined peaks indicates a relatively poor degree of crystallinity in the oxides in Fe oxide-rich microsites. Kaolinite was detected in both microsites whereas Fe oxides were detectable by XRD only in the orange microsites.

X-ray diffraction analysis of gray (upper) and orange (lower) microsites. The solid-gray symbols above the each collection of XRD spectra represent experimentally determined quartz lines using a purified quartz standard
Discussion
The hypothesis
Building upon our field and laboratory results, and on recent studies that describe coupled organic-C and Fe redox cycling in other systems (Wehrli 1990; Stumm and Sulzberger 1992; Roden et al. 2004; Thompson et al. 2006a, b), we hypothesize a potent weathering process operative in upland subsoils where redox cycles of organic C and Fe affect major restructuring of the soil environment.
The proposed weathering cycle begins with photosynthetically derived organic reductants which via rhizodeposition and organic matter’s oxidation drive reductive dissolution of Fe-(hydr)oxides and the translocation of reduced aqueous Fe away from rhizosphere and into the surrounding environment. This cycle leads to Fe oxidation and the synthesis of goethite and hematite, substantial proton flux, and the translocation, via dissolution and dispersion of aluminosilicate and weatherable minerals. Rhizospheres, periodically reduced, accumulate clay minerals that are repeatedly stripped of their Fe and other redox active metals, notably Mn, that are soluble in their reduced oxidation states. The rhizogenic origin of these redox processes represents a biological forcing of geochemical reactions along thermodynamically favorable energetics. The full hypothetical sequence is illustrated in Fig. 6, and is discussed in some additional detail in the following paragraphs.

The rhizosphere-initiated soil Fe-redox cycle, a biogeochemical system that drives mineral weathering: (a) association of rhizosphere-derived organic reductants with Fe(III)oxides in the gray microsites, (b) electron transfer to the metal oxide accompanied by the consumption of approximately 2 moles of protons per mole electron, (c) reductive dissolution of iron oxides converting Fe(III) to Fe(II) which is susceptible to hydrologic translocation in the absence of oxygen, (d) electrostatic displacement of exchangeable cations by aqueous Fe(II) ions within orange microsites, and (e) oxidative precipitation of Fe(III) oxides following re-supply of soluble oxygen generating co-adsorbed protons that promote (f) disintegration/dissolution and subsequent translocation of alumino-silicate minerals (in this case kaolinite) from orange to gray microsites
Iron oxide reduction in the absence of O2 (Eqs. 1 and 2) is a rapid process when coupled with suitable reductants, for example natural organic matter enriched in phenolic structures (Chen et al. 2003). Substantial reduction of Fe can occur on the time scale of minutes to hours (Stumm and Sulzberger 1992; Burgos et al. 2003; Chen et al. 2003; Dong et al. 2003; Chacon et al. 2006). Although anaerobic Fe reduction may be thermodynamically favorable (e.g. Eqs. 1 and 2), the rate of reduction can accelerate in the presence of reduced quinones (e.g. hydroquinones such as anthrahydroquinone-2,6-disulfonate or AHDS) (Dong et al. 2003; Chacon et al. 2006). In both abiotic and biotic Fe reduction systems, the presence of AHDS facilitates the reduction of crystalline Fe oxides and Fe-containing 2:1 layered clays, increasing both the rate and extent of Fe reduction.
Iron reduction and oxidation reactions (Eqs. 1–4) indicate favorable energetics and significant proton flux associated with Fe reductive dissolution and oxidative precipitation. We hypothesize that the oxidation of Fe(II) produces co-adsorbed protons
Reductive dissolution
Oxidative precipitation
that protonate cation exchange sites, oxy- and hydroxyl-functional groups, and promote mineral dissolution with solutes translocated away from where Fe oxidizes and into rhizospheres.
Two mineral weathering reactions (Eqs. 5 and 6) illustrate the potentially far-reaching nature of this hypothesis. Equation 5 summarizes a congruent dissolution of K-feldspar which hypothetically can occur outside of rhizospheres where Fe is oxidizing. The aqueous products of Al3+ and SiO2 diffuse or are transported by bulk flow back into rhizospheres, where they may be synthesize to kaolinite (Eq. 6). The protons generated by
kaolinite precipitation can be neutralized by the rhizosphere’s reductive dissolution of Fe (Eqs. 1 and 2). These rhizogenic reactions can hypothetically transform primary or secondary minerals in the soil or bedrock.
In addition to mineral dissolution, clays and organic matter may also be subject to dispersion during Fe–C redox cycles, which may mobilize clay into soil rhizosphere microsites. Laboratory manipulations indicate that oscillations of Fe reduction and oxidation promote dispersion and mobilization of clay colloids (Thompson et al. 2006a, b). Thompson and co-workers subjected an Inceptisol containing short-range order (SRO) Fe oxides to periodic oscillations in redox potential, oscillations that promoted precipitation of Fe oxides of increasing crystallinity, from SRO Fe oxides to hematite (Thompson et al. 2006a). During each reducing cycle, however, not only did pH increase by over a pH unit, but mineral and organic colloids were markedly dispersed (Thompson et al. 2006b). The striking textural discontinuities in these subsoil microsites will greatly benefit from further mechanistic research, as the implications are significant for the stabilization and bioavailability of carbon, nutrients, contaminant fate and transformations, and for drainage water chemistry as well.
Quantitative assessments of proton fluxes associated with Fe-redox oscillations indicate that oxidative precipitation of Fe results in substantial additions of acidity in soils. In the Calhoun subsoils, if we estimate equal volumes of gray and orange microsites within several decimeters of Bt horizon layers (Fig. 1), and if subsoils are assumed to have been uniform in Fe concentration, the ‘bulk’ soil (composite of the gray and orange microsites) would have contained 21.5 mg-Fe g−1 (Table 2). This calculation suggests that a minimum of 16.1 mg-Fe g−1 has been translocated and oxidatively precipitated away from rhizosphere microsites (21.5 plus 16.1 mg Fe g−1 equals 37.6 mg Fe g−1). Taking the molar ratio of protons produced per Fe oxidized as 2:1 (the p+/e− ratio of Eqs. 3 and 4), a total of ∼50 cmolc-H+ kg−1 can be produced within Fe-enriched volumes due to Fe oxidation. In low activity soils such as the Calhoun, in which ECEC in Fe oxide-rich microsites is only 1.7 cmolc kg−1 (Table 2), this flux of protons appears substantial relative to cation exchange charge, and has almost certainly contributed to the long-term acidification and mineral weathering in these highly weathered Ultisols. Judging from ecosystem proton budgets that have been estimated over the last 50 years, redox cycling of Fe is an underappreciated source of soil and ecosystem acidity, especially in upland soils and ecosystems (Richter and Markewitz 2001). Most terrestrial ecosystem proton budgets, including our own, (Richter and Markewitz 2001), do not even discuss the process until recently (Richter et al. 2007).
In conclusion, electron transfers between C, O2, and Fe in soil systems have been extensively described in hydromorphic soils, such as paddy-rice systems, wetlands, and aquic soils in general (Brinkman 1970; van Breemen 1988; Hardy 1993; Richardson and Vepraskas 2001; van Ranst and De Coninck 2002). This paper attempts to extend these redox reactions to environments outside of wetland soils, gleys, and pseudogleys, to upland ecosystems with deep roots that contribute organic matter to rhizospheres, and wherein reductive dissolution periodically affects redox-active metals, mineral weathering/dissolution, and colloidal mobility, all within soils that are generally well oxygenated. The evolution of spatially contrasting intra- and inter-rhizosphere microsites may be spurred by positive-feedbacks in both (1) intra-rhizosphere volumes that become increasingly fine-textured, and (2) inter-rhizosphere volumes that become increasingly coarse textured with sufficient porosity and diffusivity to promote Fe oxidation. The overall cycle of metal oxide reduction and subsequent oxidation represents an integrated biological, chemical, and physical system that may well drive mineral weathering in many upland soils.
References
Bidwell OW, Gier DA, Cipra JC (1968) Ferromagnesian pedotubules on roots of Bromus inermis and Andropogon gerardii. Cong Soil Sci Trans IV:683–692
Brinkman R (1970) Ferrolysis, a hydromorphic soil forming process. Geoderma 3:199–206
Burgos WD, Fang Y, Royer RA, Yeh G-T, Stone JJ, Jeon B-H, Dempsey BA (2003) Reaction-based modeling of quinone-mediated bacterial iron(III) reduction. Geochim Cosmochim Acta 67:2735–2748
Buerge IJ, Hug SJ (1998) Influence of organic ligands on chromium(VI) reduction by Iron(I). Environ Sci Technol 32:2092–2099
Chacon N, Silver WL, Dubinsky EA, Cusack DF (2006) Iron reduction and soil phosphorus solubilization in humic tropic forests soils: the roles of labile carbon pools and an electron shuttle compound. Biogeochemistry 78:67–84
Chan MA, Ormoe J, Park AJ, Stich M, Souza-Egipsy V, Komatsu G (2007) Models of iron oxide concretion formation: field, numerical, and laboratory comparisons. Geofluids 7:356–368
Chen J, Gu B, Royer RA, Burgos WD (2003) The roles of natural organic matter in chemical and microbial reduction of ferric iron. Sci Total Environ 301:167–178
Dierksen KP, Whittaker GW, Banowetz GM, Azevedo MD, Kennedy AC, Steiner JJ, Griffith SM (2002) High resolution characterization of soil biological communities by nucleic acid and fatty acid analyses. Soil Biol Biochem 34:1853–1860
Dong H, Kukkadapu RK, Fredrickson JK, Zachara JM, Kennedy DW, Kostandarithes HM (2003) Microbial reduction of structural Fe(III) in illite and goethtite. Environ Sci Technol 37:1268–1276
Emerson D, Weiss JV, Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants. Appl Environ Microbiol 65:2758–2761
Fimmen RL, Cory RM, Chin YP, Trouts TD, McKnight DM (2007) Probing the oxidation-reduction properties of terrestrially and microbially derived dissolved organic matter. Geochim Cosmochim Acta 71:3003–3013
Foy CD, Chaney RL, White MC (1978) The physiology of metal toxicity in plants. Ann Rev Plant Physiol 29:511–566
Glasauer S, Weidler PG, Langley S, Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium. Geochim Cosmochim Acta 67:1277–1288
Hagedorn F, Kaiser K, Feyen H, Schleppi P (2000) Effects of redox conditions and flow processes on the mobility of dissolved organic carbon and nitrogen in a forest soil. J Environ Qual 29:288–297
Hardy M (1993) Influence of geogenesis and pedogenesis on clay mineral distribution in northern Vietnam soils. Soil Sci 156:336–345
Kirk GJD (2004) The biogeochemistry of submerged soils. Wiley, Chichester, UK
Li JW, Richter DD, Mendoza A, Heine PR (2008) Four-decade responses of soil trace elements to an aggrading forest: B, Mn, Zn, Cu, and Fe. Ecology 89 (in press)
Liger E, Charlet L, van Cappellen P (1999) Surface catalysis of uranium(VI) reduction by iron(II). Geochim Cosmochim Acta 63:2939–2955
Loeppert RH, Inskeep WP (1996) Iron. In: Sparks DL (ed) Methods of soil analysis: part 3 chemical methods. Soil Science Society of America, Inc., Madison, WI, pp 639–664
Lovley DR, Giovannoni SJ, White DC, Champine JE, Phillips EJP, Gorby YA, Godwin S (1993) Geobacter metallireducens gen. nov. sp. nov., a microorganism capable of coupling complete oxidation of organic compounds to the reduction of iron and other metals. Arch Microbiol 159:336–344
Lovley DR, Coates JD, Blunt-Harris EL, Phillips EJP, Woodward JC (1996) Humic substances as electron acceptors for microbial respiration. Nature 382:445–448
Lovley DR, Fraga JL, Blunt-Harris EL, Hayes LA, Phillips EJP, Coates JD (1998) Humic substances as a mediator for microbially catalyzed metal reduction. Acta Hydroch Hydrob 26:152–157
Markewitz D, Richter DD, Allen HL, Urrego JB (1998) Three decades of observed soil acidification in the Calhoun experimental forest: has acid rain made a difference? Soil Sci Soc Am J 62:1428–1439
Markewitz D, Richter DD (2000) Long-term soil potassium availability from a Kanhapludult to an aggrading loblolly pine ecosystem. For Ecol Manage 130:109–129
Maurice PA (2002) Microbially mediated dissolution of clays: effects of siderophores. Geochim Cosmochim Acta 66:A495
Maurice PA, Lee Y-J, Hersman LE (2000) Dissolution of Al-substituted goethtites by an aerobic Pseudomonas mendocina var. bacteria. Geochim Cosmochim Acta 64:1363–1374
Mehra OP, Jackson ML (1960) Iron oxide removal from soil and clays by a dithionite-citrate system buffered with sodium bicarbonate. Clay Clay Miner 7:317–327
Michalzik B, Kalbitz K, Park J-H, Solinger S, Matzner E (2001) Fluxes and concentrations of dissolved organic carbon and nitrogen—a synthesis for temperate forests. Biogeochemistry 52:173–205
Murphy RC (1993) Mass spectrometry of phospholipids. Illuminati Press, Knoxville, TN
Neubauer SC, Emerson D, Megonigal JP (2002) Life at the energetic edge: kinetics of circumneutral iron oxication by lithotrophic iron-oxidizing bacteria isolated from the wetland-plant rhizosphere. Appl Environ Microbiol 68:3988–3995
O’Brien EL, Buol SW (1984) Physical transformations in a vertical soil-saprolite sequence. Soil Sci Soc Am J 48:354–357
Oh NH, Richter DD (2005) Elemental translocation and loss from three highly weathered soil-bedrock profiles in the southeastern United States. Geoderma 126:5–25
Oren R, Ellsworth DS, Johnsen KH, Phillips N, Ewers BE, Maier C, Schafer KVR, McCarthy H, Hendrey G, McNulty SG, Katul GG (2001) Soil fertility limits carbon sequestration by forest ecosystems in a CO2-enriched atmosphere. Nature 411:469–472
Page AL (ed) (1982) Methods of soil analysis, chemical and microbiological properties, part 2. Soil Science Society of America, Madison, Wisconsin
Qualls RG, Haines BL (1991) Geochemistry of dissolved organic nutrients in water percolating through a forest ecosystem. Soil Sci Soc Am J 55:1112–1123
Raulund-Rasmussen K, Borggaard OK, Hansen HCB, Olsson M (1998) Effect of natural organic soil solutes on weathering rates of soil minerals. Eur J Soil Sci 49:397–406
Richter DD, Markewitz D (1995) How deep is soil? BioScience 45:600–609
Richter DD, Markewitz D (2001) Understanding soil change. Cambridge University Press, UK
Richter DD, Markewitz D, Trumbore S, Wells CG (1999) Rapid accumulation and turnover of soil carbon in a re-establishing forest. Nature 400:56–58
Richter DD, Oh NH, Fimmen RL, Jackson J (2007) The rhizosphere and soil formation. In: Cardon Z, Whitbeck J (eds) The rhizosphere—an ecological perspective. Springer-Verlag pp 177–198
Roden EE, Sobolev D, Glazer B, Luther III GW (2004) Potential for microscale bacterial Fe redox cycling at the aerobic–anaerobic interface. Geomicrobio J 21:379–391
Schlesinger WH, Andrews JA (1999) Soil respiration and the global carbon cycle. Biogeochem 48:7–20
Schlichting E, Schwertmann U (eds) (1973) Pseudogley and Gley. Trans. Comm. V and VI Int. Soc. Soil Sci., Verlag Chemie, Weinheim, Germany
Schulten H-R, Schnitzer M (1998) The chemistry of soil organic nitrogen: a review. Biol Fert Soil 26:1–15
Schutter ME, Dick RP (2002) Microbial community profiles and activities among aggregates of winter fallow and cover-cropped soil. Soil Sci Soc Am J 66(1):142–153
Scott DT, McKnight DM, Blunt-Harris EL, Kolesar SE, Lovley DR (1998) Quinone moieties act as electron acceptors in the reduction of humic substances by humics-reducing microorganisms. Environ Sci Technol 32:2984–2989
Shaw JN (2001) Iron and aluminum oxide characterization for highly-weathered Alabama Ultisols. Commun Soil Sci Plan 32:49–64
Silver WL, Lugo AE, Keller M (1999) Soil oxygen availability and biogeochemistry along rainfall and topographic gradients in upland wet tropical forest soils. Biogeochemistry 44:301–328
Stone AT (1997) Reactions of extracellular organic ligands with dissolved metal ions and mineral surfaces. In: Banfield JF, Nealson KH (eds) Geomicrobiology: interactions between microbes and minerals. Mineralogical Society of America, Washington, DC, pp 309–344
Stumm W, Sulzberger B (1992) The cycling of iron in natural environments: considerations based on laboratory studies of heterogeneous redox processes. Geochim Cosmochim Acta 56:3233–3257
Thomas G (1982) Exchangeable cations. In: Page AL et al (eds) Methods of soil analysis, 2nd edn. Soil Science Society of America, Inc., Madison, WI, pp 159–165
Thompson A, Chadwick OA, Boman S, Chorover J (2006a) Colloid mobilization during soil iron redox oscillations. Environ Sci Technol 40:5743–5749
Thompson A, Chadwick OA, Rancourt DG, Chorover J (2006b) Iron-oxide crystallinity increases during soil redox oscillations. Geochim Cosmochim Acta 70:1710–1727
Trumbore S (1996) In: Boutton T, Yamasaki S (eds) Mass spectrometry of soil. Dekker, New York, pp 311–340
van Breemen N (1988) Effects of seasonal redox processes involving iron on the chemistry of periodically reduced soils. In: Stucki JW, Goodman BA, Schwertmann U (eds) Iron in soils and clay minerals. D. Reidel Publishing, Dordrecht
van Ranst E, De Coninck F (2002) Evaluation of ferrolysis in soil formation. Eur J Soil Sci 53:513–519
Vepraskas MJ (1992) Redoximorphic features for identifying aquic conditions. NC Agri Res Serv Raleigh NC Tech Bull 301, 33 pp
Vepraskas MJ (2001) Morphological features of seasonally reduced soils. In: Richardson JL, Vepraskas MJ (eds) Wetland soils. CRC Press, Boca Raton, FL, pp 163–182
Wehrli B (1990) In: Stumm W (ed) Aquatic chemical kinetics: reaction rates of processes in natural water. Wiley, New York
Weiss JV, Emerson D, Backer SM, Megonigal JP (2003) Enumeration of Fe(II)-oxidizing and Fe(III)-reducing bacteria in the root zone of wetland plants: implications for a rhizosphere iron cycle. Biogeochemistry 64:77–96
Weiss JV, Emerson D, Megonigal JP (2004) Geochemical control of microbial Fe(III) reduction potential in wetlands: comparison of the rhizosphere to non-rhizosphere soil. FEMS Microbiol Ecol 48:89–100
West, LT, Abreu MA, Bishop JP (2008) Saturated hydraulic conductivity of soils in the southern Piedmont of Georgia, USA: field evaluation and relation to horizon and landscape properties. Catena (in press)
Zhang CLL, Li YL, Ye Q, Fong J, Peacock AD, Blunt E, Fang JS, Lovley DR, White DC (2003) Carbon isotope signatures of fatty acids in Geobacter metallireductans and Shewanella algae. Chem Geol 195:17–28
Acknowledgments
The authors thank E. Vicki Hufstetler of the University of Georgia for assistance with differential X-ray diffraction analysis and differential scanning calorimetry, Paul Heine for soil analyses, Doug Dvoracek of the University of Georgia for carbon isotopic analysis, Leslie Eibest and the Duke University Biology Department for ESEM imaging, and Mr. Jim Rice for soil pit excavation. Financial support for this research was provided by the National Science Foundation (Career Grant BES-9984489 , the Programs in Biocomplexity DEB-0120698, and Long-Term Research in Environmental Biology DEB-0129383), the Andrew W. Mellon Foundation, the EPA-Science to Achieve Results (STAR), and the Sigma-Xi Grants-in-Aid-of-Research (GIAR) programs.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Fimmen, R.L., Richter, D.d., Vasudevan, D. et al. Rhizogenic Fe–C redox cycling: a hypothetical biogeochemical mechanism that drives crustal weathering in upland soils. Biogeochemistry 87, 127–141 (2008). https://doi.org/10.1007/s10533-007-9172-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-007-9172-5