
Abstract
Burkholderia sp. C3 can transform polycyclic aromatic hydrocarbons (PAHs), a class of ubiquitous pollutants, through multiple pathways, indicating existence of multiple dioxygenases (Seo et al., in Biodegradation 18:123–131, 2006a). Both phn and nag-like genes in C3 were cloned and identified with the DNA sequence alignment and the gene organization in the clusters. When cloned and expressed in Escherichia coli, either the alpha- and beta-subunits of dioxygenase of the phn genes or the ferredoxin-, alpha- and beta-subunits of the nag-like genes transformed naphthalene, phenanthrene and dibenzothiophene but at different rates. The E. coli transformant containing the phn genes transformed phenanthrene faster than that containing the nag-like genes, which was consistent with higher transcription of the phnAc gene than the nagAc-like gene in C3 in response to phenanthrene. 1-Hydroxy-2-naphthanoic acid (1H2NA) and 2-hydroxy-1-naphthanoic acid (2H1NA) (3,4- and 1,2-dioxygenation metabolites of phenanthrene, respectively) were detected in the culture medium of the phn genes transformed E. coli. The concentration of 1H2NA was 262-fold higher than 2H1NA in the medium of the phn genes transformed E. coli. The results suggested that the phn genes play a major role in 1,2-/3,4-dioxygenation and 3,4-dioxygenation dominates. Twenty-eight PAH degradation-associated enzymes including those encoded by the nag-like and phn genes in phenanthrene-grown C3 cells were identified via alignment of amino acid sequences of the detected polypeptides with those in protein databases. The polypeptides were determined with nano liquid chromatography–ion trap mass spectrometry after tryptic in-gel digestion of the enzymes on 1D SDS-PAGE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a class of important pollutants primarily from incomplete combustions. Bacteria can utilize PAHs as substrates. The most important step of PAH metabolism is the initial aromatic ring dioxygenation catalyzed by ring-hydroxylating dioxygenases (RHDs). The RHDs are commonly composed of a terminal dioxygenase iron sulfur protein and an electron transport chain. The former contains a large (α) subunit and a small (β) subunit, while the latter contains a reductase subunit and a ferredoxin subunit (Mason and Cammack 1992). Therefore, dioxygenase is a basic target for genetic screening to detect PAH-degrading bacteria (Laurie and Lloyd-Jones 2000; Moser and Stahl 2001).
Several groups of RHD genes are involved in the early step of degradation of naphthalene or phenanthrene in Gram-negative bacteria (Peng et al. 2008). In the present study, we focused on two main groups of genes, nah-like and phn genes that encode dioxygenases. The nah-like genes show a high degree of similarity (more than 90% amino acid identity) to the nah gene. The nah gene was first isolated from Pseudomonas putida strain G7 that can degrade naphthalene (Simon et al. 1993). The substrate for nah dioxygenases in P. putida is naphthalene and salicylate, while the substrate for nah dioxygenases in P. stutzeri is 2-methylnaphthalene. It was found that naphthalene and phenanthrene can be substrates for Nah dioxygenases in Comamonas testosterone GZ42 (Chauhan et al. 2008). In the present study, the isolated nah-like genes from Burkholderia sp. C3, recently isolated from PAH-contaminated soil (Seo et al. 2007), showed high sequence similarity with and gene organization similar to the nag gene cluster of Ralstonia sp. U2. The nah-like genes cloned from C3 were, therefore, called nag-like genes in the present study. Another group of genes, phn was originally isolated from Burkholderia sp. RP007 that can use naphthalene, phenanthrene, and anthracene as a sole source of carbon (Laurie and Lloyd-Jones 1999). The phn genes have a low degree of sequence similarity with the nah-like genes including nag genes. nah- and nag-like genes are mostly found in Pseudomonas and Ralstonia strains, respectively. Although multiple dioxygenase genes have been reported in some species of Cycloclasticus (Geiselbrecht et al. 1998), Mycobacterium (Moody et al. 2001), and Sphingomonas (Romine et al. 1999), to our knowledge there is no report on coexistence of nag-like and phn genes in a Burkholderia sp.
Metabolic pathways of PAHs have been well studied (Liu et al. 2010; Seo et al. 2009). Naphthalene is degraded through 1,2-dioxygenation in most bacteria (Habe and Omori 2003), whereas 2,3-dioxygenation has been reported in Bacillus thermoleovorans Hamburg 2 (Annweiler et al. 2000). Dioxygenations of phenanthrene can occur at 1,2-, 3,4- and 9,10-positions. 3,4-Dioxygenation is the major catabolic pathway of phenanthrene found in most bacteria, while some reports describe 1,2- and 9,10-dioxygenation, depending on the enzyme system found in each bacterial strain (Balashova et al. 1999; Pinyakong et al. 2000; Moody et al. 2001; Habe and Omori 2003; Seo et al. 2006b; Mallick et al. 2007). Burkholderia sp. C3 grows rapidly and can degrade phenanthrene via 3,4- and 1,2-dioxygenation (Seo et al. 2006a), while Burkholderia species were previously known to degrade phenanthrene only via 3,4-dioxygenation (Laurie and Lloyd-Jones 1999; Kang et al. 2003). The genetic study of Burkholderia sp. RP007 showed the dioxygenase enzyme encoded by phn genes responsible for 3,4-dioxygenation of phenanthrene (Laurie and Lloyd-Jones 1999). A question is then which gene encodes the enzyme that is responsible for 1,2-dioxygenation of phenanthrene in C3. The objective of the present study was to identify gene(s) and enzyme(s) that are responsible for the dioxygenations of phenanthrene in C3. In the absence of a genetic system for site-directed mutagenesis in strain C3, we applied a combination of DNA sequencing, cloning and expression in Escherichia coli, mass spectrometric analysis of metabolites, and proteomics to improve our understanding of the genes that allow strain C3 to carry out both 3,4- and 1,2-dioxygenation of phenanthrene.
Materials and methods
Bacterial strains, plasmids and culture media
The bacterial strains and plasmids used in this study are listed in Supporting Information 1 (SI1). Burkholderia sp. C3 and E. coli strains were grown in Nutrient Broth (NB) at 28°C and Luria–Bertani Broth (LB) at 37°C, respectively, until late log phase for regular cell propagation, while Minimal Medium (MM) (Bastiaens et al. 2000) was used as a basal medium for PAH degradation experiments.
Isolation of genomic DNA, plasmid DNA and DNA sequencing
Genomic DNA and plasmid DNA were isolated according to the standard procedures (Maniatis et al. 1982). DNA fragments were sequenced using a primer-walking strategy, which the primers were described in SI2.1. The ORF was identified via BLAST search to compare the DNA sequence similarity with the existing genes in NCBI database. The DNA sequences were submitted to GenBank and the accession numbers were GQ184726, GQ184727 and GQ184728 for pNag-13, pPhn-17 (part I) and pPhn-17 (part II), respectively (Fig. 1).

Schematic map and genetic organization of the isolated PAH degrading genes from Burkholderia sp. C3 on pNag-13 (a) and pPhn-17 (b). The DNA fragments were sequenced via the primer-walking strategy. Complete- and incomplete-open reading frames (ORFs) are indicated by solid-arrow and dash-arrow, respectively, and the direction of reading is indicated by the arrowheads. The relative gene designation of each ORF is indicated with italic letters. Restriction sites: B, BamHI; E, EcoRI; H, HindIII; P, PstI. The cloning regions of 2.4- and 2.0-kb into plasmid pNfb and pPab, respectively, are indicated with the vertical dash-arrows
Genomic DNA library construction and screening of cosmid containing PAH-dioxygenase genes
The total genomic DNA of Burkholderia sp. C3 was mechanically sheared to get the DNA fragments of approximately 40 kb and then ligated into pWEBTM cosmid vector (8,179 bp) (Epicentre Biotechnologies, Wisconsin, USA). A total of 1,800 clones were constructed for this library. The cosmid clones containing PAH-dioxygenase genes were screened using PCR with the primers nahAcfor and nahAcrev for the nahAc gene and the primers P8073 and P9047 for the phnAc gene (Laurie and Lloyd-Jones 2000). By using the primers nahAcfor and nahAcrev for the nahAc gene, two positive clones were found. The two positive clones had similar DNA patterns after digestion with restriction enzyme, thus the longer DNA inserted clone was selected. While using primers P8073 and P9047 for the phnAc gene, one positive clone was found.
Construction of E. coli transformants expressing dioxygenase genes
The 2.4-kb region of the ORF4, ORF5 and ORF6 encoding ferredoxin, α- and β-subunits of dioxygenase, respectively, from pNag-13 was PCR amplified as one fragment and was cloned into pGEM-T vector (Promega, Wisconsin, USA), named as pNfb (Fig. 1a) (SI2.2). The 2.0-kb region of the ORF10 and ORF11 encoding α- and β-subunits of dioxygenase, respectively, from pPhn-17 was PCR amplified as one fragment and was cloned into pGEM-T vector, named as pPab (Fig. 1b). The resulting plasmids were separately transformed into E. coli JM109, where the genes were expressed under the control of lac promoter and induction by 1 mM IPTG.
Assays for indigo formation and PAH degradation
Burkholderia sp. C3 and E. coli transformants were tested for indigo formation (Eaton and Chapman 1995) and degradation of different PAHs (SI2.3) (Seo et al. 2006a). For PAH degradation tests, the cultures (1 ml) were sampled at 0, 24, 48 and 72 h. PAHs were then extracted and analyzed with gas chromatography (GC) (SI2.4). All assays were done in triplicate, and the testing media inoculated with the E. coli containing pGEM-T vector alone and the autoclaved C3 cells were used as controls. Transformation percentages are uncorrected.
Analyses of phenanthrene metabolites
To analyze phenanthrene metabolites, C3 and the E. coli transformants were grown in the same manner as that in the PAH degradation study except the following modifications. A 250 ml aliquot of pre-grown cells was inoculated to 1 l of phenanthrene-supplemented (40 μg ml−1) MM media and incubated for 3 days. After centrifugation of the cultures, the metabolites 1-hydroxy-2-naphthoic acid (1H2NA) and 2-hydroxy-1-naphthoic acid (2H1NA) in the supernatant were extracted as previously described (Seo et al. 2006a) and analyzed on an Agilent 1200 series capillary liquid chromatograph interfaced with a Bruker high resolution time-of-flight quadrupole mass spectrometer (CapLC/micrOTOF-Q) (SI2.5). The retention times (Rt) were 16.7 min for 2H1NA and 18.1 min for 1H2NA. NESI MS of 1H2NA and 2H1NA (C11H8O3): m/z 187.056 [M–H]−, calculated, 187.040 [M–H]−. All other metabolites were previously identified and characterized with GC–MS (Seo et al. 2006a).
Quantitation of the transcription of nag-like and phn genes in C3
Total RNA was extracted with RNeasy Protect Bacteria Mini kit (Qiagen, Duesseldorf, Germany) from cultures of the C3 cells grown in MM media supplemented with 80 μg ml−1 of phenanthrene, naphthalene, 1H2NA, or 2H1NA, or 0.1% (w/v) glucose at 6, 12, 24, 36, 48, and 72 h. An aliquot of 1 μg RNA was used for reverse transcription using cDNA Reverse Transcription Kit (Applied Biosystems, California, USA). The cDNA product was diluted to 1:10 prior to the quantitative PCR (qPCR) assay, where the qPCR standards were prepared (Yin et al. 2001) and the qPCR procedure is detailed in SI2.6.
Protein extraction and analysis
To extract proteins, C3 cells were grown in MM media (1200 ml) supplemented with 80 μg ml−1 phenanthrene or 0.1% (w/v) glucose (control) for 36 h. The cells were harvested by centrifugation at 6000×g for 20 min, washed twice with 40 mM Tris–HCl (pH 7.4) and resuspended in lysis buffer [40 mM Tris–HCl, pH 7.4, 5 mM dithiothreitol (DTT), and 1 mM PMSF] containing complete protease inhibitor cocktail tablets (one tablet in 50 ml lysis buffer) (Roche Diagnostics, Mannheim, Germany). The cells were disrupted with an ultrasonic MicrosonTM cell disruptor (Misonix, New York, USA) with full power for 40 s and 1 min cooling period for ten repeated times on ice. The cell debris was removed by centrifugation at 15000×g for 40 min. The concentration of total soluble proteins from cell lysate was determined with Coomassie protein assay kit (Pierce, Illinois, USA). An approach of one dimensional gel electrophoresis coupled with LC (GE-LC) was taken to separate proteins in the GE step and tryptic peptides in the LC step followed by detection of the peptides on a Dionex UltiMateTM 3000 nano LC interfaced with a Bruker esquireHCTultra ion trap mass spectrometer (LC-ITMS) in nanoelectrospray mode. The method of SDS-PAGE and protein in-gel digestion was modified from Lee et al. (2007) as described in SI2.7.
The digested peptides were analyzed in triplicate with LC-ITMS (Lee et al. 2007). MS/MS spectra were interpreted with Mascot (Matrix Science, London, UK) via Biotools 2.2 software (Bruker); and peptide mass fingerprint (PMF) searches were performed with the Swiss-Prot and MSDB databases through the Mascot server. Peptides were assumed to be monoisotopic, oxidized at methionine residues and carbamidomethylated at cysteine residues. Up to one missed trypsin cleavage was allowed, although matches that contained any missed cleavages were not noticed. Mass tolerance was set at 1.0 Da. Probability-based molecular weight search (MOWSE) scores were estimated by comparison of search results against estimated random match populations and were reported as: 10 × log10(P), where P is the absolute probability. Scores in Mascot larger than the MOWSE score at P = 0.05 were considered statistically significant, meaning that the probability of the match being a random event is lower than 0.05. The algorithm used for determining the probability of a false-positive match with a given mass spectrum is well described by Elias et al. (2005). The false-positive rate (FPR) was estimated to be smaller than 2% [FPR = FP/(FP + TP), where FP is the number of FPR hits; TP is the number of true-positive hits]. Only proteins identified with at least two peptide hits (P ≤ 0.0025) in triplicate analyses, with each peptide containing two tryptic termini, were accepted. In addition, the MS/MS spectra of all positively identified peptides were manually confirmed twice.
Protein profiles of the treatment samples were compared with those of the appropriate control samples. Detection of a protein in the treatment sample but not in the control is referred to as up-expression/production of the protein whereas absence of a protein in the treatment sample but presence in the control is referred to as down-expression/production of the protein.
Results
Isolation of nag-like and phn gene clusters from Burkholderia sp. C3 and sequence analysis
Two clones carrying a cosmid, pNag-13 and pPhn-17, were selected from the genomic DNA library of C3. The enzymatic restriction assay revealed that DNA fragments that were ligated into pWEBTM cosmid had 35 and 33.2 kb for pNag-13 and pPhn-17, respectively. In the present study, 8417 bp were sequenced on pNag-13 and nine complete open reading frames (ORFs) were identified (Fig. 1a). The nucleotide sequences of these genes were more than 90% identity with nag genes of Ralstonia sp. U2; and the organization of these genes in the cluster was identical to that of the nag genes. The analyses of the ORFs revealed that these clusters of genes were mostly involved in the upper pathways of naphthalene catabolism (Table 1). The genes in pNag13 were, therefore, called nag-like genes. Two parts of the DNA fragment in pPhn-17 (part I, 3786 bp; part II, 7965 bp) were sequenced. Part I contained three complete and two incomplete ORFs (Fig. 1b). These proteins may be involved in the lower pathways of naphthalene catabolism, which includes transformation of salicylate via gentisate to tricarboxylic acid (TCA) cycle intermediates (Table 2). Part II contained six complete and one incomplete ORFs (Fig. 1b). These genes were 100% identity to phn genes of Burkholderia sp. RP007 and are probably related to the upper pathways of phenanthrene degradation in RP007 (Table 3). These co-existent genes imply a connection between catabolic pathways of naphthalene and phenanthrene in Burkholderia sp. C3.
Transformation of PAHs by the constructed E. coli containing nag-like and phn genes from C3
The E. coli harboring pNfb or pPab could oxidize indole to indigo. While Burkholderia sp. C3 degraded phenanthrene, naphthalene, carbazole, dibenzofuran, biphenyl, dibenzothiophene and carbaryl, the two E. coli transformants harboring pNfb or pPab transformed naphthalene, phenanthrene and dibenzothiophene (Table 4). The pPab E. coli containing the phn genes transformed phenanthrene more efficiently than the pNfb one containing the nag-like genes (89 vs. 54%). However, the pNfb E. coli transformed naphthalene completely in 2 days (data not shown), which was faster than the pPab E. coli that transformed naphthalene completely in 3 days. Both E. coli constructs transformed 64–73% of dibenzothiophene in 7 days. The results indicate the substrate preference of the enzymes.
Transcription of nag-like and phn genes in response to phenanthrene and other substrates in Burkholderia sp. C3
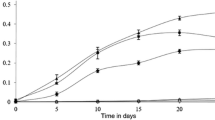
Naphthalene induced a higher level of transcription of nagAc-like gene than phenanthrene and its metabolites 1H2NA and 2H1NA since early log phase (at and before 48 h) (Fig. 2). The nagAc-like gene was significantly transcribed after mid log phase (at and after 36 h) of exposure to phenanthrene, which reached the same level as that induced by naphthalene at 72 h. The phnAc gene was transcribed at a higher level than the nagAc-like gene in response to all the substrates relative to glucose. Phenanthrene induced the transcription of phnAc gene stronger than the other substrates. Upon exposure to phenanthrene, the transcription of the phnAc gene increased gradually and reached the highest level at 48 h followed by a decrease at 72 h of culturing.

Transcription level of nagAc-like (a) and phnAc (b) genes in Burkholderia sp. C3 determined with the quantitative PCR assay at different sampling times (6, 12, 24, 36, 48 and 72 h) in response to different substrates and metabolites. Glc glucose, Phe phenanthrene, Nap naphthalene, 1H2NA 1-hydroxy-2-naphthanoic acid, 2H1NA 2-hydroxy-1-naphthanoic acid. The reactions were run in triplicate. The standard curves were constructed via running the reactions of known numbers of copies of each gene. The transcription levels were calculated as the numbers of gene copies per ng of total RNA at a specific time
phn Genes responsible for 1,2- and 3,4-dioxygenation of phenanthrene in Burkholderia sp. C3
Concentrations of the metabolites 2H1NA and 1H2NA were 0.06 and 7.55 mg l−1, respectively, in the C3 cultures (SI3). The two index metabolites were also detected in the culture of the E. coli transformant harboring pPab (phnAcAd) (ORF10 and ORF11 in pPhn-17) at concentrations of 0.004 and 1.05 mg l−1 for 2H1NA and 1H2NA, respectively, but not in that harboring pNfb. The results demonstrate that the dioxygenase α- and β-subunits encoded by phnAcAd are responsible for the 1,2- and 3,4-dioxygenation of phenanthrene although there may be also other genes and pathways operating in C3. Dioxygenases encoded by the nag-like genes may also catalyze the 1,2- and/or 3,4-dioxygenation of phenanthrene, but the degradation rate of phenanthrene would be too low to produce detectable levels of 2H1NA and 1H2NA, or the degradation rate of 2H1NA and 1H2NA would be too fast to detect them.
Differential proteomics examination of PAH-degrading enzymes in Burkholderia sp. C3 in the presence of phenanthrene and glucose
Twenty-eight proteins associated with PAH catabolism were detected in C3 in the presence of phenanthrene, but not in the presence of glucose (Table 5). Twelve of the 28 proteins could be assigned to the associated steps in the proposed phenanthrene catabolism pathways based on the functions reported in the literature (Fig. 3). The 1,2-dihydroxy-1,2-dihydronaphthalene dehydrogenase (NahB), 1,2-dihydroxynaphthalene dioxygenase (NahC) and 1,2-dihydroxybenzylpyruvate aldolase (NahE) were expressed from the nah-like gene cluster and are responsible for degradation of naphthalene analogs. Other detected enzymes include salicylaldehyde dehydrogenase (NagF) that converts salicylaldehyde to salicylic acid, salicylate hydroxylase (Nah), and catechol 1,2-dioxygenase (CatA1). The other up-expressed enzymes that directly contribute to phenanthrene degradation in C3 include dioxygenase β-subunit (PhnAd), dihydrodiol dehydrogenase (PhnB), 3,4-dihydroxyphenanthrene dioxygenase (PhnC), 2-carboxybenzaldehyde dehydrogenase (Phn), dehydrogenase (PahB), and trans-O-hydroxybenzylidenepyruvate hydratase-aldolase (PahE). In addition, proteins responsible for electron transport systems were up-expressed in C3 cells in the presence of phenanthrene relative to the cells in the presence of glucose-grown C3 cells. The results confirmed the expression of enzymes from nag-like and phn gene clusters and the multiple catabolic pathways of phenanthrene in C3.

Proposed catabolic pathways of phenanthrene and naphthalene by Burkholderia sp. C3. Genes located on pNag-13 and pPhn-17 are indicated in italic; and enzymes detected with LC-ITMS are indicated in bold in the pathways. Compounds presented in the pathways are (1) cis-3,4-dihydroxy-3,4-dihydrophenanthrene; (2) 3,4-dihydroxyphenanthrene; (3) 2-hydroxy-2H-benzo[h]chromene-2-carboxylic acid; (4) 4-(1-hydroxy-2-naphthyl)-2-oxobut-3-enoic acid; (5) 1-hydroxynaphthalene-2-carboxaldehyde; (6) 1-hydroxy-2-naphthoic acid; (7) cis-1,2-dihydroxy-1,2-dihydrophenanthrene; (8) 1,2-dihydroxyphenanthrene; (9) 3-hydroxy-3H-benzo[f]chromene-3-carboxylic acid; (10) 4-(-2-hydroxy-1-naphthyl)-2-oxobut-3-enoic acid; (11) 2-hydroxy-1-naphthaldehyde; (12) 2-hydroxy-1-naphthoic acid; (13) cis-naphthalene dihydrodiol; (14) 1,2-dihydroxynaphthalene; (15) 2-hydroxy-2H-chromene-2-carboxylic acid; (16) trans-O-hydroxybenzylidenepyruvic acid; (17) salicylaldehyde; (18) salicylic acid; (19) gentisic acid; (20) maleylpyruvic acid; (21) fumarylpyruvic acid; (22) pyruvic acid; (23) fumaric acid; (24) catechol; (25) cis,cis-muconic acid; (26) trans-2′-carboxybenzalpyruvic acid; (27) 2-carboxybenzaldehyde; (28) phthalic acid. All metabolites were identified with GC–MS (Seo et al. 2006a). In addition, the metabolites 1H2NA (6) and 2H1NA (12) were identified with capillary liquid chromatography-high resolution time-of-flight quadrupole mass spectrometry (SI2.5 and SI3)
Discussion
Co-existence of nag-like and phn genes in C3
Burkholderia sp. C3 contains nag-like and phn genes (Fig. 1). Although the multiple dioxygenase genes have been reported in various species (Geiselbrecht et al. 1998; Romine et al. 1999; Moody et al. 2001), this is the first report of identification of nag-like and phn genes in a Burkholderia sp. One question is how the two genes relate to each other and function for phenanthrene degradation in C3.
The gene sequences in the operon of cloned nag-like genes from C3 are highly similar to the nag genes. The organization of cloned nag-like genes in C3 is the same as the nag genes. The nag genes are present in Ralstonia sp. U2 and are involved in naphthalene degradation (Fuenmayor et al. 1998). The genes nagG and nagH encoding salicylate-5-hydroxylase α- and β-oxygenase components, respectively, are inserted between nagAa and nagAb that encode for ferredoxin reductase and ferredoxin of electron transport system, respectively (Fig. 1a; Table 1). This gene organization pattern is also similar to the nah operon in Comamonas testosteroni GZ42, which is responsible for naphthalene and phenanthrene degradation (Goyal and Zylstra 1997). It has been reported that the nag operon contains genes responsible for conversion of naphthalene to gentisate (Zhou et al. 2001). Moreover, ORF2 and ORF3 in pPhn-17 encode proteins similar to NagI and NagK, respectively (Fig. 1b; Table 2), which are involved in the conversion of gentisate to fumarate-pyruvate (Zhou et al. 2001). Therefore, the nag-like genes in C3 seem to play a role on catabolism of naphthalene and its structural analogs such as 1H2NA and 2H1NA.
The sequences and operon organization of the phn genes in C3 are the same as the phn operon in Burkholderia sp. RP007 (Laurie and Lloyd-Jones 1999). This phn gene may commonly exist in phenanthrene degrading Burkholderia species, while the nag-like operon in C3 might come from gene transfer. Although there is no direct evidence in the present study, the horizontal gene transfer occurs naturally among soil bacteria and plays an important role in the microbial evolution. Transfers of biodegradation genes were well proven (Matheson et al. 1996; Herrick et al. 1997; Wilson et al. 2003; Ma et al. 2006). The transposition was suggested as a major mechanism of gene transfer and rearrangement of catabolic genes in the chromosome (Herrick et al. 1997). Interestingly in the sequence of nag operon within Ralstonia sp. U2, a gene encoding a transposase is located at 1,350 nucleotides away from the end of nag operon (nagN) (Zhou et al. 2001). Thus, genes from nag operons could possibly be transferred from Ralstonia sp. U2 to other soil bacteria through the mechanism of transposition.
PAH degradation by nag-like and phn genes encoded RHDs in C3
Although RHDs are multi-component enzymes, the recombinant dioxygenases of nag-like and phn genes from C3 in this study can transform PAHs in the transformed E. coli even the recombinant dioxygenase lack of reductase-subunit in pNfb or lack of both ferredoxin- and reductase-subunit in pPab (Fig. 1; Table 4). Simon et al. (1993) suggested that E. coli contain a non-specific electron transport system, which could function as the ferredoxin- and reductase-subunit for the recombinant dioxygenase. The nag-like and phn encoded RHDs from C3 in the E. coli transformants can completely degrade naphthalene within 3 days and 64–73% of dibenzothiophene in 7 days (Table 4). The E. coli transformants possessing Nag- and Phn-dioxygenases transformed 54 ± 10 and 89 ± 6% of phenanthrene, respectively, in 3 days. It is shown that dibenzothiophene-degrading enzymes encoded by the dox-gene cluster, isolated from Pseudomonas sp. C18, also degrade naphthalene and phenanthrene (Denome et al. 1993). This indicates that the dioxygenase is a multi-substrate enzyme. Although the sequence of α-subunit of the terminal dioxygenase iron sulfur proteins, NagAc (nagAc) (ORF5 in pNag-13) and PhnAc (phnAc) (ORF10 in pPhn-17), from C3 did not show high similarity with each other, they can catalyze the transformation of naphthalene, phenanthrene, and dibenzothiophene. It is reported that the conserved His208, His213 and Asp362 are involved in the catalytic site of the RHD (Denome et al. 1993). Asn201, Phe202, Phe352, Asp205, Val260, Trp316, Trp358 and Thr351 have been reported to be involved in substrate binding at the catalytic site of the enzyme (Kauppi et al. 1998; Parales et al. 2000). The three amino acids at the catalytic site are present, although at different positions, in the α-subunits NagAc (His206, His211 and Asp362) and PhnAc (His209, His214 and Asp365) of C3. The eight amino acids involving substrate binding remain in their positions relative to the positions of the three catalytic site amino acids in C3 (SI4). This suggests that both NagAc and PhnAc of C3 can bind PAHs and catalyze the dioxygenation of PAHs such as naphthalene, phenanthrene and dibenzothiophene. Parales et al. (2000) showed that the α-subunit of dioxygenase determines the substrate specificity through some specific amino acids. Although 53% of the amino acids are identically aligned between NagAc (238 of 448 aâ) and PhnAc (238 of 451 aâ) from C3 (SI4), the difference probably affects the substrate pocket or active site of the enzymes, signifying the substrate preference and catalysis efficiency between the two types of dioxygenase. The nag-like and phn genes of C3 can be an alternative degradation route for each other, or perhaps co-responsible for the dioxygenation of naphthalene, phenanthrene and dibenzothiophene. Dioxygenase encoded by phn genes can efficiently transform phenanthrene when compared with that encoded by nag-like genes isolated from C3.
phn Genes are responsible for 1,2- and 3,4-dioxygenation of phenanthrene in C3
To our knowledge, this is the first report describing that phnAcAd are responsible for 1,2-dioxygenation of phenanthrene. In general, dioxygenases derived from the phn gene cluster of Burkholderia species catalyze 3,4-dioxygenation (Laurie and Lloyd-Jones 1999; Kang et al. 2003). However, metabolites from 1,2- and 3,4-dioxygenations of phenanthrene were found in C3 culture media (Seo et al. 2006a). Based on the results of DNA cloning and expression of phn genes in E. coli, the PAH transformation analysis, and mass spectrometry analysis of the metabolites, we feel that these evidences demonstrate that PhnAcAd (dioxygenase α- and β-subunits) are responsible for the two pathways (Fig. 3; Table 4, SI3), although 3,4-dioxygenation, producing the metabolite 1H2NA, dominates because the concentration of 1H2NA was 128-fold of 2H1NA. Both 1H2NA and 2H1NA were detected in the culture of the E. coli transformant although their formation from phenanthrene requires additional enzymes beyond the Phn dioxygenases. The E. coli host probably has the necessary enzymes to catalyze further degradation of dihydrodiol products to 1H2NA or 2H1NA. The metabolites 1H2NA and 2H1NA inside the cells are metabolized to naphthalene-1,2-diol and then to phthalic acid or salicylic acid. Salicylate is then metabolized via gentisate degradation pathway (Seo et al. 2006a).
Expressions and functions of nag-like and phn genes in C3 relevant to phenanthrene catabolism
The results of DNA cloning, gene expression and metabolite analysis strongly support that the phn genes are mainly responsible for dioxygenation of phenanthrene. The nag-like genes may contribute partially to phenanthrene catabolism because the E. coli transformant harboring the nag-like genes degraded 54 ± 10% of phenanthrene in 72 h (Table 4). The nag-like genes probably play major roles on catabolism of phenanthrene metabolites, which was supported by (a) undetectable levels of 1H2NA and 2H1NA in the culture of the transformed E. coli containing pNfb and (b) high levels of the nagAc-like gene transcription in response to naphthalene and phenanthrene in C3 (Fig. 2). Several naphthalene-analogous metabolites (4–6, 10–12, and 14 in Fig. 3) are produced during phenanthrene catabolism (Seo et al. 2006a). These metabolites inside the cells may induce the transcription of the nagAc-like gene. The upper pathway of phenanthrene catabolism in C3 would operate through enzymes encoded by the phn gene cluster (Part II of pPhn-17 in Fig. 1b) (Fig. 3). A cluster of the nag-like genes on pNag-13 (ORF1-9) is similar to the nag operon, which is probably responsible for catabolism of naphthalene to gentisate; and the nag-like genes in Part I of pPhn-17 (ORF1-5) are probably responsible for converting gentisate to pyruvic acid (Tables 1, 2). The high transcription of nagAc-like gene at 48–72 h in the presence of phenanthrene may be coincident with the catabolism of metabolites that are structurally similar to naphthalene and are derived from phenanthrene degradation (Fig. 2). Both the nag-like and phn genes were not highly transcribed in the wild C3 cells in response to 1H2NA and 2H1NA. This is likely related to probable poor absorption of the two metabolites into the cells because of the high polarity of their molecules (Figs. 2, 3).
Up-expression of multiple operons during phenanthrene catabolism
Comparison of the protein profiles in the phenanthrene-fed and glucose-fed C3 cells supports the genetic and catabolic data of phenanthrene catabolism. The term of up-expression or up-regulation used here means the proteins that were detected and identified only in the phenanthrene-fed cells, but not in the glucose-fed cells. The proteins such as PhnAd and PhnB involved in phenanthrene dioxygenation are up-expressed in the phenanthrene-fed C3 cells. The enzymes catalyzing naphthalene transformation are probably involved in the lower pathways of phenanthrene catabolism. PhnAd was detected and identified although the α-subunit (PhnAc) was not detected. This may be due to many factors including the limit of detection, sample preparation, trypsin digestion, LC and MS conditions, and post-translational modifications.
Among the differentially up-expressed enzymes include NahB, NahC, NahE, NagF, and various electron transport proteins, confirming the corresponding involvement of multiple operons during the phenanthrene catabolism. Other PAH-degrading enzymes, such as Bph and Dox for biphenyl and dibenzothiophene catabolism, respectively, are up-regulated in the phenanthrene-fed cells. This is consistent with the ecological environment, i.e., PAH-contaminated soil where C3 is isolated and it is adapted to utilize a variety of PAHs. The presence of one PAH, thus, would readily induce multiple PAH degrading genes some of which may be involved in catabolism of other related PAHs.
This study shows the co-existence of nag-like and phn gene clusters in Burkholderia sp. C3. The two types of dioxygenases degrade naphthalene, phenanthrene and dibenzothiophene in varying efficiencies. The phn genes play more dominant roles than nag-like genes on the initial dioxygenations of phenanthrene. The dioxygenase encoded by phnAcAd is primarily responsible for both 1,2- and 3,4-dioxygenation of phenanthrene whereas the dioxygenases encoded by nag-like genes mainly govern the lower catabolic pathways in C3.
Abbreviations
- PAHs:
-
Polycyclic aromatic hydrocarbons
- RHD:
-
Ring-hydroxylating dioxygenase
- 1H2NA:
-
1-Hydroxy-2-naphthanoic acid
- 2H1NA:
-
2-Hydroxy-1-naphthanoic acid
References
Annweiler E, Richnow HH, Antranikian G, Hebenbrock S, Garms C, Franke S et al (2000) Naphthalene degradation and incorporation of naphthalene-derived carbon into biomass by the thermophile Bacillus thermoleovorans. Appl Environ Microbiol 66:518–523
Balashova NV, Kosheleva IA, Golovchenko NP, Boronin AM (1999) Phenanthrene metabolism by Pseudomonas and Burkholderia strains. Process Biochem 35:291–296
Bastiaens L, Springael D, Wattiau P, Harms H, deWachter R, Verachtert H, Diels L (2000) Isolation of adherent polycyclic aromatic hydrocarbon (PAH)-degrading bacteria using PAH-sorbing carriers. Appl Environ Microbiol 66:1834–1843
Chauhan A, Faziurrahman, Oakeshott JG, Jain RK (2008) Bacterial metabolism of polycyclic aromatic hydrocarbons: strategies for bioremediation. J Ind Microbiol 48:95–113
Denome SA, Stanley DC, Olson ES, Young KD (1993) Metabolism of dibenzothiophene and naphthalene in Pseudomonas strains: complete DNA sequence of an upper naphthalene catabolic pathway. J Bacteriol 176:2158–2164
Eaton RW, Chapman PJ (1995) Formation of indigo and related compounds from indolecarboxylic acids by aromatic acid-degrading bacteria: chromogenic reactions for cloning genes encoding dioxygenases that act on aromatic acids. J Bacteriol 177:6983–6988
Elias JE, Haas W, Faherty BK, Gygi SP (2005) Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2:667–675
Fuenmayor SL, Wild M, Boyes AL, Williams P (1998) A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J Bacteriol 180:2522–2530
Geiselbrecht AD, Hedlund BP, Tichi MA, Staley JT (1998) Isolation of marine polycyclic aromatic hydrocarbon (PAH)-degrading Cycloclasticus strains from the Gulf of Mexico and comparison of their PAH degradation ability with that of Puget Sound Cycloclasticus strains. Appl Environ Microbiol 64:4703–4710
Goyal AK, Zylstra GJ (1997) Genetics of naphthalene and phenanthrene degradation by Comamonas testosterone. J Ind Microbiol Biotechol 19:401–407
Habe H, Omori T (2003) Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotechnol Biochem 67:225–243
Herrick JB, Stuart-Keil KG, Ghiorse WC, Madsen EL (1997) Natural horizontal transfer of a naphthalene dioxygenase gene between bacteria native to a coal tar-contaminated field site. Appl Environ Microbiol 63:2330–2337
Kang H, Hwang SY, Kim YM, Kim E, Kim YS, Kim SK et al (2003) Degradation of phenanthrene and naphthalene by a Burkholderia species strain. Can J Microbiol 49:139–144
Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S (1998) Structure of an aromatic ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6:571–586
Laurie AD, Lloyd-Jones G (1999) The phn genes of Burkholderia sp. strain RP007 constitute a divergent gene cluster for polycyclic aromatic hydrocarbon catabolism. J Bacteriol 181:531–540
Laurie AD, Lloyd-Jones G (2000) Quantification of phnAc and nahAc in contaminated New Zealand soils by competitive PCR. Appl Environ Microbiol 66:1814–1817
Lee SE, Seo JS, Keum YS, Lee KJ, Li QX (2007) Fluoranthene metabolism and associated proteins in Mycobacterium sp. JS14. Proteomics 7:2059–2069
Liu M, Cui Y, Duan Y, Zhong J, Sun W, Wang M et al (2010) Synthesis of metabolites of polycyclic aromatic hydrocarbons. Mini Rev Org Chem 7:134–144
Ma Y, Wang L, Shao Z (2006) Pseudomonas, the dominant polycyclic aromatic hydrocarbon-degrading bacteria isolated from Antarctic soils and the role of large plasmids in horizontal gene transfer. Environ Microbiol 8:455–465
Mallick S, Chatterjee S, Dutta TK (2007) A novel degradation pathway in the assimilation of phenanthrene by Staphylococcus sp. strain PN/Y via meta-cleavage of 2-hydroxy-1-naphthoic acid: formation of trans-2,3-dioxo-5-(29-hydroxyphenyl)-pent-4-enoic acid. Microbiology 153:2104–2115
Maniatis T, Fritsch EF, Sambrook J (1982) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Mason JR, Cammack R (1992) The electron-transport proteins of hydroxylating bacterial dioxygenases. Annu Rev Microbiol 46:277–305
Matheson VG, Forney LJ, Suwa Y, Nakatsu CH, Sexstone AJ, Holben WE (1996) Evidence for acquisition in nature of a chromosomal 2,4-dichlorophenoxyacetic acid/a-ketoglutarate dioxygenase gene by different Burkholderia spp. Appl Environ Microbiol 62:2457–2463
Moody JD, Freeman JP, Doerge DR, Cerniglia CE (2001) Degradation of phenanthrene and anthracene by cell suspensions of Mycobacterium sp. strain PYR-1. Appl Environ Microbiol 67:1476–1483
Moser R, Stahl U (2001) Insights into the genetic diversity of initial dioxygenases from PAH-degrading bacteria. Appl Microbiol Biotechnol 55:609–618
Parales RE, Lee K, Resnick SM, Jiang H, Lessner DJ, Gibson DT (2000) Substrate specificity of naphthalene dioxygenase: effect of specific amino acids at the active site of the enzyme. J Bacteriol 182:1641–1649
Peng RH, Xiong AS, Xue Y, Fu XY, Gao F, Zhao W et al (2008) Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev 32:927–955
Pinyakong O, Habe H, Supaka N, Pinpanichkarn P, Juntongjin K, Yoshida T et al (2000) Identification of novel metabolites in the degradation of phenanthrene by Sphingomonas sp. strain P2. FEMS Microbiol Lett 191:115–121
Romine MF, Stillwell LC, Wong KK, Thurston SJ, Sisk EC, Sensen C et al (1999) Complete sequence of a 184-kilobase catabolic plasmid from Sphingomonas aromaticivorans F199. J Bacteriol 181:1585–1602
Seo JS, Keum YS, Hu Y, Lee SE, Li QX (2006a) Degradation of phenanthrene by Burkholderia sp. C3: initial 1,2- and 3,4-dioxygenation and meta- and ortho-cleavage of naphthalene-1,2-diol. Biodegradation 18:123–131
Seo JS, Keum YS, Hu Y, Lee SE, Li QX (2006b) Phenanthrene degradation in Arthrobacter sp. P1-1: initial 1,2-, 3,4- and 9,10-dioxygenation, and meta- and ortho-cleavages of naphthalene-1, 2-diol after its formation from naphthalene-1,2-dicarboxylic acid and hydroxyl naphthoic acids. Chemosphere 65:2388–2394
Seo JS, Keum YS, Harada RM, Li QX (2007) Isolation and characterization of bacteria capable of degrading polycyclic aromatic hydrocarbons (PAHs) and organophosphorus pesticides from PAH-contaminated soil in Hilo, Hawaii. J Agric Food Chem 55:5383–5389
Seo JS, Keum YS, Li QX (2009) Bacterial degradation of aromatic compounds. Int J Environ Res Public Health 6:278–309
Simon MJ, Osslund TD, Saunders R, Ensley BD, Suggs S, Harcourt A et al (1993) Sequences of genes encoding naphthalene dioxygenase in Pseudomonas putida strains G7 and NCIB-9816-4. Gene 127:31–37
Wilson MS, Herrick JB, Jeon CO, Hinman DE, Madsen EL (2003) Horizontal transfer of phnAc dioxygenase genes within one of two phenotypically and genotypically distinctive naphthalene-degrading guilds from adjacent soil environments. Appl Environ Microbiol 69:2172–2181
Yin JL, Shackel NA, Zekry A, McGuinness PH, Richards C, Putten KVD et al (2001) Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) for measurement of cytokine and growth factor mRNA expression with fluorogenic probes or SYBR Green I. Immunol Cell Biol 79:213–221
Zhou NY, Fuenmayor SL, Williams PA (2001) nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. J Bacteriol 183:700–708
Acknowledgments
This work was supported in part by grants from Hawaii State Civil Defense, Hawaii Department of Agriculture Pesticides Branch, the US-EPA award 989512-01-1, USDA TSTAR awards, US ONR NRL award N00173-05-2-C003, and the ONR HEET award N00014-09-1-0709.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Tittabutr, P., Cho, I.K. & Li, Q.X. Phn and Nag-like dioxygenases metabolize polycyclic aromatic hydrocarbons in Burkholderia sp. C3. Biodegradation 22, 1119–1133 (2011). https://doi.org/10.1007/s10532-011-9468-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10532-011-9468-y