
Abstract
Containing the spread of established invasive species is critical for minimizing their ecological impact. Effective containment requires sensitive sampling methods capable of detecting new introductions when invaders are at low density. Here we explore whether environmental DNA (eDNA) sampling could be used as a surveillance tool to detect new incursions of aquatic invasive species on offshore islands. We develop an eDNA molecular assay for invasive cane toads (Rhinella marina) in Australia, validate our assay on the mainland, and apply it to an offshore island (Moreton Island) that is a target of ongoing cane toad surveillance. Our eDNA assay correctly identified four mainland sites at which cane toads were observed, as well as a fifth site within 1 km of known populations. Five additional sites outside the cane toad’s current distribution tested negative for cane toad eDNA. Site occupancy detection models indicated that two water samples and three qPCR replicates were sufficient to achieve a cumulate detection probability > 0.95. Applying our eDNA assay to samples from 19 sites on an offshore island over a 2-year period revealed the absence of cane toad eDNA, in line with our current understanding of cane toad distribution. Our results suggest that eDNA sampling could be strategically applied to meet the Australian Commonwealth’s objective of maintaining cane toad-free offshore islands.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Once a non-native species becomes established in a new area, the likelihood of successful eradication markedly declines. Thus, in many cases, the most cost-efficient approach to minimize the ecological impact of established invaders is to contain their spread (Epanchin-Niell and Hastings 2010). Effective containment requires ongoing monitoring to ensure that new incursions are detected before a species can successfully establish. Environmental DNA (eDNA) sampling—detection of a species’ extracellular DNA in environmental samples—is a sensitive monitoring method capable of detecting species at low densities (Pilliod et al. 2013; Smart et al. 2015; Dougherty et al. 2016); eDNA sampling could, therefore, act as an early warning signal for new incursions ahead of invasion fronts, or on offshore islands (Xia et al. 2018; Tingley et al. 2017).
Globally, offshore islands provide important refuges for taxa threatened by invasive species on mainland areas. In Australia, offshore islands host some of the last remaining strongholds for populations of species that have declined on the mainland due to widespread invasive species, such as the cane toad, Rhinella marina. Consequently, in its cane toad threat abatement plan, the Australian Commonwealth identified the maintenance of cane toad-free offshore islands as a key management priority (Commonwealth of Australia 2011). To achieve that aim, we urgently need sensitive and cost-efficient monitoring methods that can be applied to areas at risk of invasion, such as transport hubs and tourist destinations.
In this note, we develop an eDNA quantitative PCR (qPCR) assay for detecting the presence of R. marina on offshore islands. We validate this assay on known mainland R. marina populations and apply it to an offshore island frequented by tourists and therefore at risk of R. marina invasion.
Methods
Sample collection: eDNA validation
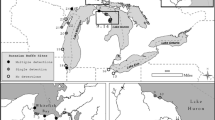
In April 2017, we validated our eDNA assay at five separate mainland waterbodies in the Clarence Valley region of coastal northern New South Wales (Yamba, Angourie, and Mororo) that were within the current distribution of R. marina. Adult R. marina were observed at four of those sites during water sample collection; tadpoles were observed at three sites. We also evaluated the specificity of our eDNA assay at five waterbodies that were between 20 and 650 km from the known distribution of toads in New South Wales (Shark Creek, Avoca, Taren Point, and Nowra; Fig. 1a). Both permanent and ephemeral waterbodies were sampled; waterbodies were between ~ 75 and 1500 m3. At each site, we took three 20–240 mL water samples with Hapool sterilized disposable syringes (Shandong Hapool Medical Technology, China). Water was drawn from the site with the syringe and passed through an attachable (leur lock) Sterivex® 0.22 μm filter unit (Merck, Germany). Variation in water volumes filtered through the Sterivex® units was largely due to the filters clogging. These filters are an efficient DNA capture method (Spens et al. 2016), despite increased risk of clogging. Samples were collected from approximately equidistant locations around the edge of each water body. Filter units were stored on ice in a dark storage container following collection, and shipped to The University of Melbourne, Victoria for processing. Once at the laboratory, samples were stored at − 20 °C until DNA extraction could occur.

a Breeding season length for cane toads as predicted by a ecophysiological model (Kearney et al. 2008). The dark line demarcates the toad’s distribution as of 2017 (Tingley et al. 2017). The hollow stars on the mainland show the locations at which eDNA samples were collected to validate the eDNA assay. The black circle and arrow on the east coast show the location of Moreton Island, where the eDNA assay was applied. The inset b shows the approximate locations at which eDNA samples were collected on Moreton Island
Sample collection: eDNA application
We applied our eDNA assay to 19 waterbodies on Moreton Island, Queensland, a large offshore island 25 km from the mainland (Fig. 1b). Rhinella marina colonised the closest large mainland city (Brisbane) in 1945. Importantly, Moreton Island lies within the cane toad’s fundamental niche (Fig. 1; Kearney et al. 2008).
Moreton Island has two main access points: Wrecks campground (− 27.162595°, 153.370517°) and Tangalooma Island Resort (− 27.181140°, 153.371733°). Live and dead adult R. marina have been found occasionally on the island, most likely due to hitch-hiking on camping gear from visitors. Dead R. marina have also been observed washed up on beaches. Since 2013, Brisbane City Council have deployed R. marina scent detection dogs on several occasions each year for detecting R. marina incursions and/or breeding on the island. While the scent detection dogs have detected several R. marina adults, to date there has been no evidence of breeding on the island, and no groups of cane toads have been observed.
eDNA sampling on Moreton Island was undertaken on two occasions (December 2016 and May 2017) and was targeted at waterbodies where the scent detector dogs had given an indication of R. marina presence but visual searches had revealed no R. marina individuals. Eleven sites were sampled in December 2016 over 2 days, and 12 sites were sampled in May 2017 over 2 days. Four of the sites sampled in 2017 were also sampled in 2016. Samples were taken as above, with 60–300 mL water passed through the Sterivex® filter units.
Probe development, DNA extraction and qPCR analysis
Species-specific primers and a TaqMan® minor groove binding (MGB) probe were developed for R. marina to target an 80 bp fragment spanning part of the mitochondrial tRNA-Gly and NADH dehydrogenase subunit 3 (ND3) genes; there is only one haplotype of this region known to be present in Australia (Slade and Moritz 1998). The primers and probe were ordered as a custom TaqMan® gene expression assay from Life Technologies; forward primer R.Marina_ND3_F ACCCCAGGAGAAAATAATGTCTCT, reverse primer R.Marina_ND3_R ACCAGAAGCTAACAGTGGCTAAAAT, MGB probe R.Marina_ND3_MGB CAATTGCTAGGGTAATAAA. Primer specificity was checked using a Blast search of the NCBI nucleotide database, with no close matches found outside of R. marina.
DNA was extracted from the filters using the Qiagen DNeasy Blood & Tissue Kit (using the spin-column protocol). 540 μL of ATL buffer and 40 μL of proteinase K were added to each filter unit. Each filter was then sealed and incubated at 56 °C for 3 h with constant agitation. The lysis solution was transferred into new 2 mL tubes. Hereafter the Qiagen DNeasy Blood & Tissue Kit manufacturers protocol was followed for the remaining part of the DNA extraction, with the following minor adjustments: 500 μL AL buffer, 500 μL ethanol, and final elution step of 100 μL AE buffer for each sample.
Real-time TaqMan® PCR assays were conducted using a Roche LightCycler 480 system in a 384-well format. 10 μL reactions containing 5 μL of 2 × Qiagen multiplex PCR Master Mix (Qiagen), 0.5 μL 20 × TaqMan® Gene Expression Assay, 2.5 μL ddH2O, and 2 μL of DNA were prepared in triplicate. Included in each 384-well assay plate were control reactions containing 10, 1, 0.1 0.01 0.001, 0.0001 ng of R. marina DNA and a negative control with no DNA template. The amplification occurred in conditions of 15 min at 95 °C, followed by 15 s at 95 °C and 1 min at 60 °C for 50 cycles. The amplification profiles of each PCR were used to determine the crossing point (Cp) value using the Absolute Quantification module of the LightCycler® 480 software package. A TaqMan® Exogenous Internal Positive Control VIC probe was run for each sample to test for the presence of PCR inhibitors. No inhibition was detected. The qPCR reaction efficiency calculated from the DNA standard was 100%, while the R2 value was 0.99.
All extractions and qPCR analysis were undertaken in a room that is dedicated to low-quantity DNA sources. Negative controls were included at all stages (DNA extraction, qPCR) so that contamination issues could be identified if present. No contamination was detected. A sample was considered positive if 1/3 qPCR replicates detected the target DNA.
Site occupancy detection modelling
To analyse eDNA data from the five sites within the toad’s distribution, we used a site occupancy detection framework. Our model was defined by a sequence of Bernoulli trials:
where \(z_{i}\) describes the latent presence (\(z_{i}\) = 1) or absence (\(z_{i}\) = 0) of toad eDNA at site i given the probability of occupancy \(\psi\); and \(a_{ij}\) denotes the presence (\(a_{ij}\) = 1) or absence (\(a_{ij} = 0)\) of toad eDNA in water sample j from site i, as a function of the occurrence of eDNA, \(z_{i}\), and the availability probability \(\theta\). The observed detection data \(y_{ijk}\) are a function of the occurrence of eDNA in water sample j from site i, and the probability of detecting eDNA in quantitative PCR (qPCR) replicate k, \(\omega\) (Schmidt et al. 2013; Lugg et al. 2018).
We estimated conditional cumulative availability probabilities (\(\theta^{ *}\)) and qPCR detection probabilities (\(\omega^{*}\)) for eDNA sampling, using mean estimates of eDNA availability probability (\(\theta\)), and qPCR detection probability (\(\omega )\) respectively, estimated from the site occupancy detection model.
where \(\theta^{ *}\), and \(\omega^{*}\) are conditional cumulative probabilities of detection following K water samples/qPCR replicates, respectively (Schmidt et al. 2013).
Models were fit in R v3.2.1 (R Core Team 2016) using JAGS v3.4.0 (Plummer 2003) and the R2jags package (Su and Yajima 2015). Three Markov chains were each run for 50,000 iterations, discarding the first 20,000 iterations of each chain. Prior distributions for \(\theta\) and \(\omega\) were specified as logistically distributed with location = 0 and scale = 1 on the logit scale. The prior distribution for \(\psi\) was specified as logistically distributed with location = 1.5 and scale = 0.5 on the logit scale (with higher weight given to high probabilities of occurrence), to account for our prior knowledge of toad occurrence at the five sites. Trace plots indicated chains were well mixed; all R hat values were ~ 1.0.
Results and discussion
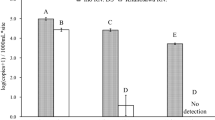
Our eDNA assay detected the presence of R. marina eDNA at all four mainland sites at which toads were visually observed (average concentrations at each site were 5 pg/L, 14 pg/L, 632 pg/L and 1707 pg/L). Consequently, site occupancy detection models estimated a high probability of occupancy (Mean \(\psi\) [95% credible interval] = 0.812 [0.431, 0.998]). Importantly, mean estimates of eDNA availability at the water sample level (\(\theta\) = 0.857 [0.606, 0.991]) and of probability of detection at the qPCR level (\(\omega\) = 0.731 [0.566, 0.867]) suggested that two water samples and three qPCR replicates (the number of replicates used here) were sufficient to achieve a cumulative detection probability > 0.95 (Fig. 2). Thus, eDNA sampling appears to be a highly sensitive tool for detecting cane toad populations.

Cumulative availability probabilities a and qPCR b detection probabilities of eDNA sampling for cane toads Rhinella marina at five sites on the Australian mainland. Boxes show medians (dark lines) with interquartile ranges. The broken horizontal line represents a cumulative detection probability = 0.95
Two out of three water samples from a fifth mainland site where toads were not visually detected also tested positive for toad eDNA (1/3 and 2/3 qPCR replicates, with a mean concentration of 6 pg/L). While false positive detections are always a possibility with environmental samples (Lahoz-Monfort et al. 2016), several lines of evidence suggest that this was not the case here. First, additional qPCR replicates run on these two positive samples also detected the species’ eDNA (1/3 samples with 2/3 qPCR replicates). Second, none of the DNA extraction blanks or qPCR blanks successfully amplified R. marina eDNA, suggesting that laboratory contamination of samples was unlikely. Third, three replicate samples from each of five sites outside the current distribution of R. marina tested negative for toad eDNA. The probability of any one of those five sites being occupied, given the observed detection histories (000), was very low: (1 − \(\theta\))3 = 0.00299. Finally, this fifth mainland site was within 1 km of known R. marina populations, suggesting that the species may have indeed colonised this location but not been visually detected. Collectively, these results suggest that our eDNA assay is not only sensitive but also specific. Nonetheless, we recommend that new detections (e.g., ahead of invasion fronts or on islands) be interrogated by running additional qPCRs on positive samples, and/or by collecting additional water samples from the site(s) in question, prior to development of a formal management response.
Our assay revealed a lack of R. marina eDNA at high-risk areas frequented by tourists on Moreton Island, which is currently thought to be free of R. marina. High probabilities of detection on mainland Australia, coupled with ongoing surveillance efforts on the island, suggest that this island indeed lacks established cane toad populations. The eDNA assay developed here could be applied to additional ongoing surveillance programs in high-risk areas where R. marina is currently absent, or where eradication efforts are underway. The eDNA assay may be particularly useful on islands in northern Australia that harbour predators that are sensitive to the toad’s toxin, such as northern quolls (Dasyurus hallucatus). Our assay could also be applied to islands throughout the Caribbean and Pacific where cane toads have established non-native populations.
Our results contribute to a growing body of evidence that eDNA sampling is a highly sensitive technique for monitoring the spread of invasive species (Dejean et al. 2012; Smart et al. 2015; Adrian-Kalchhauser and Burkhardt-Holm 2016; Dougherty et al. 2016). Judicious application of eDNA sampling to high-risk transport hubs, such as air and sea ports or tourist areas, could provide an effective means with which to contain invader spread and protect key biodiversity assets.
References
Adrian-Kalchhauser I, Burkhardt-Holm P (2016) An eDNA assay to monitor a globally invasive fish species from flowing freshwater. PLoS ONE 11:e0147558. https://doi.org/10.1371/journal.pone.0147558
Commonwealth of Australia (2011) Threat abatement plan for the biological effects, including lethal toxic ingestion, caused by cane toads. Department of Sustainability, Environment, Water, Population and Communities, Canberra, Australia
Dejean T, Valentini A, Miquel C et al (2012) Improved detection of an alien invasive species through environmental DNA barcoding: the example of the American bullfrog Lithobates catesbeianus. J Appl Ecol 49:953–959. https://doi.org/10.1111/j.1365-2664.2012.02171.x
Dougherty MM, Larson ER, Renshaw MA et al (2016) Environmental DNA (eDNA) detects the invasive rusty crayfish Orconectes rusticus at low abundances. J Appl Ecol 53:722–732. https://doi.org/10.1111/1365-2664.12621
Epanchin-Niell RS, Hastings A (2010) Controlling established invaders: integrating economics and spread dynamics to determine optimal management. Ecol Lett 13:528–541. https://doi.org/10.1111/j.1461-0248.2010.01440.x
Kearney M, Phillips BL, Tracy CR et al (2008) Modelling species distributions without using species distributions: the cane toad in Australia under current and future climates. Ecography 31:423–434. https://doi.org/10.1111/j.0906-7590.2008.05457.x
Lahoz-Monfort JJ, Guillera-Arroita G, Tingley R (2016) Statistical approaches to account for false-positive errors in environmental DNA samples. Mol Ecol Resour 16:673–685. https://doi.org/10.1111/1755-0998.12486
Lugg WH, Griffiths J, van Rooyen AR et al (2018) Optimal survey designs for environmental DNA sampling. Methods Ecol Evol 9:1049–1059. https://doi.org/10.1111/2041-210X.12951
Pilliod DS, Goldberg CS, Arkle RS et al (2013) Estimating occupancy and abundance of stream amphibians using environmental DNA from filtered water samples. Can J Fish Aquat Sci 70:1123–1130. https://doi.org/10.1139/cjfas-2013-0047
Plummer M (2003) JAGS: A program for analysis of Bayesian graphical models using Gibbs sampling. Proc Int Workshop Distrib stat Comput 124:125
R Core Team (2016) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Schmidt BR, Kery M, Ursenbacher S et al (2013) Site occupancy models in the analysis of environmental DNA presence/absence surveys: a case study of an emerging amphibian pathogen. Methods Ecol Evol 4:646–653. https://doi.org/10.1111/2041-210X.12052
Slade RW, Moritz C (1998) Phylogeography of Bufo marinus from its natural and introduced ranges. Proc Royal Soc B: Biol Sci 265(1398):769–777
Smart AS, Weeks AR, van Rooyen AR et al (2015) Environmental DNA sampling is more sensitive than a traditional survey technique for detecting an aquatic invader. Ecol Appl 25:1944–1952
Spens J, Evans AR, Halfmaerten D et al (2016) Comparison of capture and storage methods for aqueous macrobial eDNA using an optimized extraction protocol: advantage of enclosed filter. Methods Ecol Evol 8:635–645. https://doi.org/10.1111/2041-210X.12683
Su Y-S, Yajima M (2015) R2jags: using R to Run ‘JAGS’. R package version 0.5-7. https://CRAN.R-project.org/package=R2jags
Tingley R, Ward-Fear G, Schwarzkopf L et al (2017) New Weapons in the Toad Toolkit: A review of methods to control and mitigate the biodiversity impacts of invasive cane toads (Rhinella marina). Q Rev Biol 92:123–149. https://doi.org/10.1086/692167
Xia Z, Zhan A, Gao Y et al (2018) Early detection of a highly invasive bivalve based on environmental DNA (eDNA). Biol Invasions 20:437–447. https://doi.org/10.1007/s10530-017-1545-7
Acknowledgements
RT and ARW were funded by the Australian Research Council Linkage Scheme (LP140100731). MG would like to thank Sutherland Shire Council and Sydney Metropolitan Local Land Services for funding, and Trent McKenna and George Madani for assistance in the field.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tingley, R., Greenlees, M., Oertel, S. et al. Environmental DNA sampling as a surveillance tool for cane toad Rhinella marina introductions on offshore islands. Biol Invasions 21, 1–6 (2019). https://doi.org/10.1007/s10530-018-1810-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-018-1810-4