
Abstract
Alien predators exert severe effects on island ecosystems, and their eradication from island habitats may therefore be necessary to conserve the native biota. Efforts are being made to eradicate the American mink (Neovison vison) from the Atlantic Islands of Galicia National Park (NW Spain), a protected site inhabited by vulnerable island fauna. We applied a molecular genetic approach to elucidate the source of the invaders and to evaluate the effectiveness of the trapping programme. We collected mink scats in the field and obtained tissue samples from culled mink. Populations of feral mink were known to be present in coastal areas close to the National Park archipelagos in the 1980–1990s. However, the molecular findings suggest that these populations were not the main source of the mink populations that colonized the islands during the 2000s. Recent releases from farms directly on to the islands are a more likely source of these invaders. Genetic analysis suggested that mink reproduced successfully on the islands and were relatively isolated from other mainland populations. The findings also suggest that most of the culled mink were juveniles, probably because it was more difficult to catch adults. Since mink are short-lived animals, it seems that eradication may also be achieved when a large proportion of juveniles are culled in isolated and small populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alien species are considered one of the most serious threats to biodiversity worldwide (Pimentel 2011; Funk 2015). Islands are particularly vulnerable to invasion by non-native species because, commonly, insular biota evolve under low predation pressures and they often lack effective methods of defence against non-natural enemies (Reaser et al. 2007). Alien predators, especially mammals, have a particularly severe impact on native island fauna often leading to the extinction of entire populations of native-species (Courchamp et al. 2003; Blackburn et al. 2004), with subsequent cascades of effects on ecosystems (Simberloff 2011; Medina et al. 2014). The eradication of alien predators from islands is increasingly used as a management strategy to conserve island biota (Keitt et al. 2011; Dawson et al. 2015).
Genetic bottlenecks and demographic stochasticity are likely to occur when low numbers of founders settle in a new area (Simberloff 2009), which have negative effects on invasion success (Tobin et al. 2011; Bock et al. 2015). However, genetic admixture due to multiple or continuing sources of invasive individuals may have positive consequences for the fitness and the adaptive potential of founder groups, thus enhancing their chances of persistence, especially in novel habitats (Dlugosch and Parker 2008; Facon et al. 2008; Zalewski et al. 2010). Thus, the successful eradication of alien vertebrates from islands depends on the initial number and composition of the founder group (Dlugosch and Parker 2008), as well as the degree of connectivity to mainland populations (Lockwood et al. 2009).
Molecular genetic analyses have been particularly useful for clarifying the dynamics of invasion (Lawson Handley et al. 2011; Fitzpatrick et al. 2012), and also for guiding management actions (Robertson and Gemmell 2004; Le Roux and Wieczorek 2009; Fraser et al. 2013). Analyses of nuclear genetic diversity and genetic structure (i.e. genetic differentiation among populations) are particularly helpful for identifying source populations and routes of invasion, which is critical for determining methods and units of eradication or control (Robertson and Gemmell 2004; Abdelkrim et al. 2005; Adams et al. 2014). For example, when invading populations cluster with one of the potential source populations and show reduced genetic diversity a stepping stone scenario of invasion (i.e. serial colonization) may be inferred (Lawson Handley et al. 2011; Estoup and Guillemaud 2010). On the other hand, kinship-based methods and genetic networks may be especially valuable for studying population dynamics and migration rates in recently diverged populations (Rollins et al. 2012; Veale et al. 2013). Potential immigrants may be recognized as non-related individuals in a genetic network (Rollins et al. 2012).
The American mink (Neovison vison; hereafter “mink”), a semi-aquatic carnivorous mammal, is an established alien species with one of the greatest impacts on vertebrate diversity in Europe, South America and Asia (Macdonald and Harrington 2003; Bonesi and Palazón 2007). Native to North America, mink were introduced to many European countries for fur farming. Escapes from farms gave rise to feral populations, with dramatic impacts on several species of native vertebrates (Dunstone 1993; Bonesi and Palazón 2007). Mink are highly mobile (Gerell 1970) and are able to swim relatively long distances (more than 2.5 km) and they can reach off-shore islands by island hopping (Björsson and Heirstensson 1991). Accidental boat transportation may also facilitate the colonization of some remote islands (Ratcliffe et al. 2008). Mink commonly have a devastating impact on the native biota on islands, particularly on seabirds (Craik 1997; Nordström et al. 2002). Eradication of invasive species from small coastal islands may be a successful strategy (Roy et al. 2015), especially when potential recolonization is taken into account (Dawson et al. 2015).
In Galicia (NW Spain), the first free-living American mink populations were detected in the 1970s, close to the sites of fur farms (Delibes 1983; Vidal and Delibes 1987). Despite the continual expansion of the species across Galicia (see also Rodrigues et al. 2015), the large islands remained free of mink until the early 2000s (Pereira 2006). The Atlantic Islands of Galicia National Park comprises a group of islands (Fig. 1) that are protected with the aim of conserving island biota, including several species of vulnerable fauna. The presence of mink was first detected in two groups of islands (Cíes and Sálvora) in 2003, and dramatic predation events on native fauna were later recorded, e.g. on endangered seabirds (Velando and Munilla 2011; Barros et al. 2014). As a result, after some early control attempts, the National Park established a culling programme in 2009.

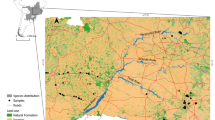
Study area showing the location of the Atlantic Islands of Galicia National Park and coastal locations where mink abundance was estimated by surveying line transects (black lines on the coastline). Grey circles indicate scat presence in June 2014. No mink have been recorded on the Island of Ons. The minimum distances between coastline and islands with mink populations are shown
Here, we carried out a retrospective analysis of the invasion process and of the efforts made to eradicate the American mink from Atlantic Islands of Galicia National Park. The aims of this study were twofold: (1) to analyse the possible sources of invasive populations and (2) to evaluate the success of the culling programme. Thus, in order to elucidate the source of invaders, we genotyped mink by using hair samples from culled animals and faecal samples collected in the National Park and adjacent mainland areas. Using data collected during the culling program and kinship-based methods, we also reconstructed mink population dynamics in the National Park. We suggest that this approach may be useful for examining the effectiveness of trapping programmes and evaluating successful strategies for eradication of invasive predators from islands.
Materials and methods
Study area
This study was carried out in the Atlantic Islands of Galicia National Park, a group of islands in Galicia (NW Spain, Fig. 1). Mink are known to have colonized two of these islands, Sálvora and Cíes, as early as 2003 (see Results). The small Island of Sálvora (190 ha) is located in the Ría de Arousa, close to the Barbanza peninsula (c. 3 km), and surrounded by a large number of islets (Fig. 1), so mink could reach Sálvora by island hopping (Björsson and Heirstensson 1991). The Cíes Islands form an archipelago (434 ha) located at the mouth of the Ría de Vigo, close to the Morrazo peninsula (c. 2.5 km) and Val Miñor coast (c. 5 km). The Cíes archipelago comprises three islands: San Martiño and the twin islands of Monteagudo and Faro, connected by a sandy beach and a rocky bridge. The islands of San Martiño and Faro are separated by a small strait (A Porta, c. 500 m).
We first gathered evidence for the presence of mink close to the study area (see Supplementary Material [SM]). In June 2014, we established transects in continental coastal areas close to the National Park (see Fig. 1) in order to obtain samples from possible sources and estimate mink abundance. We surveyed the following areas (from north to south): Corrubedo-Aguiño (Barbanza area); Grove-Faxilda (Salnés area); Udra-Cangas (Morrazo area) and Monteferro-Silleiro (Miñor area), all potential sources from mainland populations surrounding the National Park archipelagos. A total of 11 coastal line transects (30.5 km) were sampled. The length of each transect varied between 1 and 5 km (2.8 ± 1.3 km, see Fig. 1). Mink abundance in coastal transects was estimated as the number of scats per km (see above).
Sampling and DNA amplification
Hair samples were collected by the National Park staff from frozen samples of some culled mink and preserved in ethanol (25 samples in Sálvora and 2 samples in Cíes). Due to logistic problems, the mink could not be stored for a long time and, unfortunately, their sex and age were not determined before disposal. Additionally, we collected scats in surveys in coastal areas (see above) and in the National Park (see below). The scats were preserved in ethanol (Cíes: 7 scats, Sálvora: 62 scats, coastal areas: 123 scats).
DNA was extracted from scat samples by using the Qiamp DNA Stool Kit (Qiagen, Netherlands) according to the manufacturer’s instructions. DNA was extracted from the hair samples obtained from culled mink by a standard phenol:chloroform method. After a preliminary screening of several microsatellites, we selected six microsatellite loci that have previously been developed for mustelids (Mvi57, Mvi111, Mv219 and Mvi232, O’Connell et al. 1996; Mvis002 and Mvis075, Fleming et al. 1999), and their amplification showed consistent results. Amplifications were carried out in two multiplex polymerase chain reactions (PCRs) performed in a GeneAmp PCR System 2700 thermal cycler (Applied Biosystems, see details in SM).
Genetic diversity and private alleles
For all microsatellites analyzed, departures from Hardy–Weinberg equilibrium (HWE) were tested from estimated values of Wright’s F, according to Weir and Cockerham (1984), by using GENEPOP Version 3.3 (Raymond and Rousset 1995). The Markov chain-randomization procedure (Guo and Thompson 1992) was used to calculate p values. Dememorization number, number of batches, and number of iterations per batch were set at 1000. As the number of private alleles, i.e. alleles found only in a single population among a group of populations, strongly depends on sample size, we used a rarefaction approach as implemented in ADZE software (Szpiech et al. 2008). Under a single source scenario, we expected a smaller number of private alleles than in multiple founder populations.
Genetic structure and genetic assignment
The genetic differentiation (F ST) between all pairs of populations was calculated using a randomization procedure to determine the significance, according to Weir and Cockerham (1984). To visualize the patterns of differentiation among populations, we performed a factorial correspondence analysis on the genotypes for all the loci/alleles. The analysis was run using the Genetix 4.05.02 software (Belkhir et al. 2004). Conventionally, the first axis makes a larger contribution to total inertia and usually displays the differentiation between major population groups. We also generated a neighbour-joining (NJ) tree based on Nei’s standard genetic distance (D ST; Nei 1972) using the POPTREEW software (Takezaki et al. 2014). The tree was midpoint rooted, and levels of confidence in tree nodes were calculated from 100,000 bootstrap replications.
We assessed the genetic structure of samples by using two different clustering techniques. We applied the CLUSTER_DIST, a software for clustering individuals (Rodríguez-Ramilo et al. 2009), to assess the genetic structure. In CLUSTER_DIST, the method is implemented using a simulated annealing algorithm to find the partition that possesses the maximal average genetic distance between subpopulations. Simulated annealing is an optimization procedure that is suitable for dealing with many genetic problems. Parameters used to run the CLUSTER_DIST software in this study included 100,000 alternative solutions generated per step, a maximum number of steps (temperatures) set to 400, and an initial temperature (T) of 0.001, which was reduced during each step by a factor of Z (cooling factor) equal to 0.9. The possible number of populations evaluated (K) ranged from two to ten and ten replicates were run for each K. The number of inferred clusters was estimated by an approach that follows the same rationale as Evanno et al. (2005), but adapted to genetic distances. It was based on the rate of change in the averaged genetic distance between successive K values (ΔK) calculated as ΔK = |D(K + 1) − 2D(K) + D(K − 1)|, where D was the averaged genetic distance in the optimal solution for a given K. The inferred number of clusters corresponds to the value with the highest ΔK (see Rodríguez-Ramilo et al. 2009 for more details). The Bayesian clustering method in STRUCTURE yielded rather similar results (data not shown).
We assigned island samples to the most likely location of genetic source by using the Bayesian assignment test method in GENECLASS2 (Piry et al. 2004). We used Rannala and Mountain (1997) computation criteria by including the mainland populations (see Fig. 1) as reference populations. We also estimated the assignment probability (see Piry et al. 2004) by 10,000 Montecarlo simulations using the algorithm proposed by Paetkau et al. (2004).
Culling and scat surveys
Systematic culling programmes aimed at eradicating or controlling mink populations were established by the National Park between 2009 and 2014, although some sporadic trapping had been carried out since 2005. Mink were live-trapped in individual cage metal traps with a single entrance mainly placed on the coastline (see details in SM). The culling campaigns took place on Sálvora and the Cíes Islands between October and April each year (Table 1). Additionally, between 2007 and 2014, as part of the control programme, the relative abundance of mink in the National Park was estimated by recording the number of scats in coastline transects (Table 1; see also details in SM,). The results were grouped into two main periods (see SM for details), the breeding season (February–June) and non-breeding season (July–January). Capture rate and mink abundance were analyzed by General Linear Models (GLM), assuming a Gaussian distribution of errors, with island and season as fixed factors and year as covariate.
Genetic network
Relatedness (r) for all possible pairwise combinations of individuals within each island population (Sálvora and Cíes) was estimated by the maximum likelihood approach (Wagner et al. 2006), using the ML-Relate software (Kalinowski et al. 2006). In accordance with sampling events, we classified mink samples into seasons, following the mink reproductive cycle (see above; years: 2009–2013, from July year t to June year t + 1). Relatedness in all individuals captured in the same season was compared by a GLM assuming a Gaussian distribution and with island and season as independent factors.
We examined the genetic relationships between individuals within island populations by building a genetic network (Rollins et al. 2012) on pairwise coancestry values. These values were estimated using the MOL_COANC software (Fernández and Toro 2006), which implements an estimator belonging to the group of ‘pedigree reconstruction’. Due to the lack of information about the age of individuals and their sex (in most cases) a simplified analysis was performed by assuming all sampled individuals were contemporary (i.e. the same generation). A single generation pedigree was simulated and, thus, estimable relationships are equivalent to full-sibs, half-sibs or non-related animals. The number of available ‘virtual’ parents in the reconstructed genealogy was set to the maximum sensible value (i.e. one father and one mother for each individual). We ran five solutions for each island population and pairwise coancestry was estimated as the most probable relationship in these solutions. For mink genotyped in each island, coancestry relationships were visualized with NETDRAW v2.148 (Borgatti 2002). Networks were built with the spring embedding algorithm, which cluster densely connected nodes and displace less connected nodes to the edge. We used genetic networks to investigate family groups and also to detect candidate immigrants, such as those genetically dissimilar to other island members and hence possibly originating outside the island (Rollins et al. 2012). Thus, we identified possible candidate immigrants as those individuals with no coancestry across all possible dyads and/or no coancestry in the same or previous sampling event. Additionally, a parental sibship was reconstructed for each island by using full likelihood method implemented in COLONY v2.0.5.8 (Wang 2009, 2013; Jones and Wang 2010) to estimate the effective population size (Ne) and to infer some reproductive parameters (see details in SM).
Population modelling
Management strategies for eradicating American mink populations were assessed by population modelling of a mink population with the average parameters derived from the literature. The main objective of the modelling was to determine which combination of population parameters in this baseline population might help to remove mink from relatively isolated populations, such as those in islands, and to compare these results with the observed population dynamics in the Atlantic Islands of Galicia National Park. We developed an age-classified model (Caswell 2001) based on mink life-history traits (Dunstone 1993) by using a female model with two age classes (juveniles and adults, see details in SM). Note that we were interested in comparing different scenarios (see below), rather than quantifying the population dynamics. We simulated the effect on extinction probability (i.e., proportion of runs where the population went extinct) by running Monte Carlo simulations (1000 runs) over time (12 years) using PopTools (Hood 2010), under three different extreme-case scenarios: (1) no culling and different number of colonizers (propagule pressure, 2–20 mink), (2) culling only adult mink, (3) culling only juvenile mink. In all scenarios, we also evaluated the effect of immigration rate (0–8 annual immigrants) on extinction probability.
Results
Mink presence in coastal areas
Since the early 1970s, mink have been released to the wild as a result of farm closures, accidental escapes or deliberate releases (see SM, Fig S1). In 1992, feral mink were already present in the Rías Baixas, close to the Atlantic Islands of Galicia National Park (Fig S1e), and twenty-years later, feral mink were widely distributed across Galicia (Fig S1f).
In June 2014, mink scats were especially abundant (6.2 ± 1.9 scats km−1) in the coastal areas closest to the Cíes Islands (Morrazo and Miñor, Fig S2). Mink were present on the Barbanza coast (1.8 ± 1.0 scats km−1), close to the Island of Sálvora, but no scats were found on the Salnés coast (Fig S2).
Microsatellite variation
Overall, 210 samples were used for genetic analysis (Table S1). The observed and expected heterozygosity (Ho and He) varied among loci and to a lesser extent among populations (SM, Table S1). Overall, mink populations showed similar mean number of alleles per locus (corrected by sample size), except Barbanza, where allelic richness was very low (Fig. S3a). The mean number of private alleles per locus, as functions of standardized sample sizes, showed that mink from Cíes and Sálvora had a larger number of private alleles than mink from coastal sites (Fig. S3b).
Population genetic structure
The level of genetic differentiation (FST) between all pairs of sampled mink populations is shown in Table 2. The Sálvora population was strongly differentiated (the highest FST value) from Barbanza population, the closest mainland population. By contrast, the Sálvora population was more similar to the Cíes population (FST = 0.097). Additionally, the Cíes and Miñor populations were also poorly differentiated. Interestingly, the populations from Miñor and Morrazo yielded the lowest FST value, suggesting a large exchange of individuals between mainland populations in the Ría de Vigo (see Fig. 1).
The first three axes of the Factorial Correspondence Analysis (FCA) explained around 95% of the observed variability between mink genotypes (Fig. 2a). The first axis of the factorial correspondence analysis (53% of variance) separated island populations (Sálvora and Cíes), with positive values, from mainland populations, with negative values. The second and third axes mainly separated respectively the Cíes and Miñor populations from other populations. The population-level tree also discriminated, with relatively high bootstrap support, (92%) island and mainland populations (Fig. 2b). In the mainland node, the node separating Barbanza population had low support (62%).

Population genetic structure of American Mink in southwest Galicia. a Loadings (mean ± SD) of sampled populations on the first three axis (percentage of explained variance shown in parenthesis) extracted by a factorial correspondence analysis using Genetix v. 4. b Population-level neighbour-joining tree based on Nei’s standard genetic distance. Percent bootstrap support (100,000 replicates) shown at nodes. c Hidden genetic structure obtained by a clustering analysis (using CLUST_DIST) for K = 2–4 clusters. Individuals were grouped by sampled populations for comparison
The optimal number of clusters determined suggested that model with three genetic clusters (K = 3) was considerably better than the other models. For this and larger number of clusters, a cluster (pale brown in Fig. 2c) was represented in all sampled populations, probably indicating a shared genetic background. A cluster (dark green in Fig. 2c) broadly corresponded to Sálvora and some Cíes individuals. Additionally, more than a half of the individuals from Miñor and Morrazo were clustered in the same deme (brown in Fig. 2c), which also included three individuals from Cíes. Clustering analysis suggested that the Barbanza population was very homogenous, with all individuals clustered together even when K = 5.
Genetic assignment of individuals from island
Overall, the probability of mink from the National Park being assigned to mainland populations as the source of colonization was low (mean ± SD, 0.16 ± 0.24; Fig. 3). The probability of individuals from Sálvora being assigned to Barbanza, the closest mainland population, was not different from zero (0.017 ± 0.069). Similarly, the probability of most mink sampled in Sálvora being assigned to any mainland population was very low, except for a single sample that was unexpectedly assigned to the most distant population (Miñor, Fig. 3). Although higher than for Sálvora, the probability that the Cíes specimens were founded by mainland populations was also low (Fig. 3). Assignment to Miñor showed the highest, although also low, probability (0.32 ± 0.29), with two individuals showing a relatively high probability (>0.90) of being assigned to Miñor.

Heat map of assignment probability of individual mink samples from islands to coastal populations estimated by GeneClass2. Each column represents one individual sample (column width differs according to sample size: Sálvora N = 31, Cíes N = 64) and colour indicates the probability of being assigned to a reference population
Culling and mink dynamics in island populations
In total, 90 minks were culled in the National Park (43 minks on the Island of Sálvora and 47 on the Cíes Islands) between 2005 and 2014. In the systematic culling campaigns, the capture rate (minks per trapping days) decreased over time (r = −0.61, F 1,12 = 7.49, P = 0.018; Fig. 4a–d), but was similar between islands (F 1,12 = 0.40, P = 0.54). On the Island of Sálvora, capture rates were higher in the non-breeding than in the breeding season (Fig. 4c), although the differences were not significant (F 1,3 = 1.22, P = 0.35). On the Cíes Islands, the highest capture rate was recorded in the 2010 breeding season (Fig. 4d), but seasonal differences were not significant (F 1,3 = 0.79, P = 0.40).

Mink in the Atlantic Islands of Galicia National Park: Top panels a, b show the total number of mink caught per year during systematic culling campaigns. Middle panels c, d show the annual number of mink caught per trapping days. Bottom panels e, f show the annual mink abundance (scats per km) estimated in line transect surveys (see "Methods"). Left panels present results from Sálvora island and right ones correspond to Cíes islands
The highest numbers of mink scats were recorded in 2009 in the non-breeding season (Sálvora, 2.5 scats km−1; Cíes 6.6 scats km−1). Overall, the results of scats surveys suggested a decrease in mink abundance over time on both islands (r = −0.53, F 1,21 = 8.35, P = 0.009, Fig. 4e–f) and higher abundances on Cíes than on Sálvora (F 1,21 = 9.91, P = 0.005, Fig. 4e–f). Seasonal differences in mink abundance were not significant on either island groups (F 1,21 = 2.63, P = 0.12). On the Cíes Islands, marked differences between the non-breeding and breeding season in 2009 and 2011 (Fig. 4f) may suggest a high reproductive output in these years, although overall seasonal differences were not significant (F 1,9 = 1.66, P = 0.23). No signs of mink presence have been recorded since 2013 on the Island of Sálvora, and since 2014 on the Cíes Islands.
Genetic network in island populations
Mink samples collected in the same season (from July to June of the following year) showed similar distributions of pairwise relatedness values on both islands (mean relatedness ± SD: Sálvora: 0.27 ± 0.03; Cíes: 0.25 ± 0.03; F 1,1246 = 0.46, P = 0.50).
The genetic network built on coancestry values (equal or higher than half-sibs) revealed dense connected nodes on both islands (Fig. 5a). Thus, most individuals (97%) had at least one relative among the sampled individuals. Only one mink from Sálvora and two mink from Cíes did not have first-order genetic relationships with other mink sampled on the same island. In the samples from Sálvora, one mink was also not related to any other in the same or previous sampling events (Fig. 5a). Importantly, genetic networks reconstructed five full-sib families with 2.8 ± 0.37 (mean ± SE) members on Sálvora and ten full families with 3.4 ± 0.6 members on Cíes (Fig. 5b).

Genetic network of mink populations from the islands of Sálvora (left panels) and Cíes (right panels). a Genetic network built on first-order genetic relationships (coancestry equal or higher than half-sibs). Nodes represent individual samples, colours indicate the annual sampling event and lines connect dyads sharing a genetic relationship. Arrows indicate individuals that were not genetically related to any other individual in the same or previous sampling event, i.e. candidate immigrants. b Families of first-order genetic relationships (full siblings and/or parent-offspring)
The estimated reproductive parameters suggested successful reproduction in several seasons on both islands (Table S3). Effective population size, estimated by the COLONY full likelihood method, was higher on Cíes (29) than on Sálvora (17). Only two culled mink from Sálvora were identified as breeders (Table S3).
Island population modelling
In a mink population with average parameters derived from the literature, we found that the initial number of invaders had an effect on extinction probability only under conditions of no immigration; this effect disappeared with continuous immigration (Fig S4a). As expected, the eradication of mink from islands is achieved by trapping fewer individuals when culling adults (Fig. S4b) rather than juveniles (Fig S4c). Overall, culling juveniles is a worse strategy compared with culling adults. Nevertheless, our model suggested that the eradication goal could also be achieved when a large proportion of juveniles are culled in small and closed populations (Fig S4c). This is because mink are short-lived mammals with few reproductive events (Bonesi et al. 2006).
Discussion
In this study, we investigated the invasion of American mink on Atlantic Islands of Galicia National Park, a protected insular area in NW Spain, and evaluated the outcome of the mink eradication programme. Populations of feral mink were already present in coastal areas close to the National Park archipelagos in 1980–1990s, probably as escapes from fur-farms, when mink farming was poorly developed. In 2003, mink were first detected in two of the island groups (Sálvora and Cíes) in this National Park (Pereira 2006). In 2009, direct and indirect observations revealed a high abundance of mink on these islands, similar to that observed in nearby coastal areas.
Sources of invasive populations
In the Ría de Vigo, there was a low level of differentiation between Miñor and Morrazo, suggesting a homogeneous genetic structure in this area, but both were genetically differentiated from Barbanza, the most distant population. Landscape features in Galicia, such as mountains and Rías, probably limit gene flow among populations (Lecis et al. 2008; Zalewski et al. 2009, 2010; Fraser et al. 2013). These populations were already established in the 1980–1990s, as a result of escapes from small farms with no breeding management. Thus, coastal populations probably originated from small inbred lines, which may have been mostly subjected to natural selection and genetic drift (Lee 2002; Dlugosch and Parker 2008).
Genetic analyses consistently showed that the Sálvora and Cíes populations were closely related. Overall, these results suggested that islands were colonized by a common mink pool and argued against a simple stepping-stone model of island colonization from nearby feral mink populations. Founder effects such as reduced diversity and spatial structure (Austerlitz et al. 1997; Le Corre and Kremer, 1998) are expected in the stepping stone-model. Nevertheless, island mink showed a high degree of genetic diversity, which is similar to or even higher than that of mainland mink. The number of private alleles in the island populations exceeded those in coastal populations, suggesting that island mink are not merely a subsample of mainland populations (Szpiech and Rosenberg 2011).
The mink from Sálvora were genetically differentiated from the Barbanza population, geographically the closest feral population (Fig. 1). Furthermore, genetic assignment by Genclass also suggested that the Sálvora population did not originate from this nearby population. In Barbanza, we found few mink scats, and the microsatellites analysis indicated a very low degree of genetic diversity, suggesting a marginal and small population. Otters are common in this coastal area and may prevent mink settlement (Bonesi and Macdonald 2004; McDonald et al. 2007).
The mink population in the Cíes islands showed a small genetic signature from a nearby mainland population (Miñor). Interestingly, two individuals from Sálvora and two individuals from Cíes showed a high probability (>85%) of being assigned to the Miñor population. It is possible that Miñor was the source population that colonised Cíes and thereafter Sálvora. Given the distance between Sálvora and both Miñor and Cíes (open water distance, >30 km; coastal line distance, >140 km), and the absence of mink from Island of Ons (situated between Cíes and Sálvora islands), the possibility of exchange of numerous individuals between these populations seems unlikely. Incidental boat transportation cannot be entirely ruled out (Manchester and Bullock 2000), although this has not been documented in our study area.
A possible scenario is that recent releases from farms directly on to the islands were the initial source leading to island invasion. Mink can rapidly establish populations after escaping from farms (Bonesi and Palazón 2007). From the mid 1990s, mink breeding programmes were established in Galicia, and farm owners annually exchanged animals for breeding. This cross-breeding strategy aimed at enhancing genetic diversity probably led to genetic homogenization among mink farms (e.g. Michalska-Parda et al. 2009). During the 2000s, releases from fur farms produced a large number of potential invaders. Recent populations originating from this farm pool could create a genetic structure where mink from distant new populations are genetically closer than populations from adjacent sites, but established decades ago (see Zalewski et al. 2010). Thus, the genetic differentiation between island and mainland populations may reflect genetic differences between feral and farm-reared mink at the time of island invasion (e.g. Thirstrup et al. 2015).
Culling and eradication
Ninety mink were culled in the National Park and the capture rate declined over time in both archipelagos. The annual decrease in mink abundance was revealed by scat surveys and capture rates, indicating that the culling programme was successfully reducing mink population in both areas. Indeed, at this time the National Park is apparently free of mink: no signs of their presence (photos from camera traps, scats, footprints, carcasses of depredated animals, etc.) were recorded in extensive surveys carried out during 2015 and 2016. In addition to other successful mink culling programs (Moore et al. 2003; Nordström et al. 2002), removal of mink from the National Park confirms that mink eradication is a feasible task, especially on coastal islands.
The removal effort was rather similar between periods, but the trapping rate was higher during the non-breeding period (except on Cíes in 2010). In the parentage assignment analysis, only one captured male was assigned as a breeder, suggesting that most trapped mink were juveniles. It is possible that some adults avoid traps (Zuberogoitia et al. 2006) or that their home range was outside the trapping area. Traps were not placed in some areas because of logistic constraints and difficult topography. Overall, the results suggest that trapping was successful for capturing autumn dispersing juveniles (Bonesi and Palazón 2007), but probably only captured a very small proportion of adults. Nevertheless, these results should be taken with caution because not all culled individuals were genotyped.
Demography and model of invasion
The findings of the genetic network analysis suggest that island mink populations were relatively isolated during the study period. Only two mink from each island were candidate immigrants (individuals non-genetically connected to previous generations; Rollins et al. 2012), and only one individual from Sálvora was genetically related to mink captured in later sampling events. Thus, some immigrants may have reached the islands, but the contribution of immigration to mink demography on the islands was (if any) very low (see Fraser et al. 2015).
The genetic network also suggests successful reproduction after island colonization. Indeed, kits were observed during mink surveys. Reproduction may be more successful on Cíes than on Sálvora, as revealed by parental analyses but also by the differences in scat abundance between non-breeding and breeding seasons. Rocky islands, such as Cíes and to a lesser extent Sálvora, may show exceptionally high mink densities (see Roy 2011). Overall, our results suggest that islands were colonised by a relatively diverse group of animals in some early events. These mink were relatively isolated, but reproduced successfully. Thus, after an initial colonization period, mink seem to be able to establish a large viable population in adequate novel habitats with no enemies, such as rocky islands of the National Park, even with negligible gene flow.
The population model developed supports the idea that a relatively small number of colonizers may establish a viable population, even in the absence of immigration (but under no inbreeding depression, see Forsman 2014). Our model suggests that eradication is clearly more efficient when culling is focussed on adults than juveniles. Reduced mink density in response to trapping may also increase female reproduction (Melero et al. 2015), which may even exacerbate the differences between the two trapping scenarios in achieving the eradication goal. Nevertheless, as shown by here, adults may be difficult to catch, but juveniles are highly mobile and trappable during autumn dispersal (see Roy 2011). American mink are relatively short-lived mammals (Bonesi et al. 2006, 2007; Hammershøj et al. 2005). Capturing animals (including juveniles) prior to the reproductive season could be an alternative strategy in terms of cost-effectiveness, although only when the proportion of culled individuals is large and populations are relatively isolated (see Anderson et al. 2016). When there is no immigration, culling may also reduce mink populations to a small critical size in which inverse density dependent effects (i.e., Allee effects) are likely to occur (Courchamp et al. 2008).
At the present, mink seems to have been eradicated from Atlantic Islands of Galicia National Park. The results of this study guided management strategy, which thus mainly focused on culling within the islands, rather than mainland trapping. The successful removal of mink from the National Park has probably played a crucial role in improving the conservation status of endangered seabirds (Barros et al. 2016). Nevertheless, a major risk of eradication failure on islands is the ability of the invasive species to recolonize. In our study, we detected some candidate immigrants, but with minor genetic impact on populations. Nevertheless, in mink-free islands, the opportunity increases for newcomers, so the likelihood of rapid reinvasion from nearby mink populations should be not underestimated (Fraser et al. 2015). An early detection system and rapid trapping response to invasion must be maintained. The latter should involve collection of samples (tissue or scats) when mink are detected in order to determine whether they are survivors or re-invaders (see Veale et al. 2012).
Escapes from farms are an important source of potential colonizers. Thus, effective measures, such as building secure fences and placing mink traps around the farms, must be implemented. In addition, genotyping farmed mink may help to trace and control the pathways of invasion. Mink populations in the mainland areas close to the National Park are probably spatially differentiated, which implies that each mink population may be a management unit. Further exhaustive identification of genetically differentiated management units in mainland areas may guide control efforts in these areas (Bifolchi et al. 2010; Fraser et al. 2013; Zalewski et al. 2009). Interestingly, mink populations in peninsulas, such as those surrounding the National Park, may be managed as islands if gene flow is minimised by, for example, culling campaigns during autumn dispersal. From this study, we learned that mink eradication is possible, even with low adult capture rates, when immigration is prevented.
References
Abdelkrim J, Pascal M, Calmet C, Samadi S (2005) Importance of assessing population genetic structure before eradication of invasive species: examples from insular Norway rat populations. Conserv Biol 19:1509–1518
Adams AL, van Heezik Y, Dickinson KJM, Robertson BC (2014) Identifying eradication units in an invasive mammalian pest species. Biol Invasions 16:1481–1496
Anderson DP, McMurtrie P, Edge KA, Baxter PWJ, Byrom AE (2016) Inferential and forward projection modelling to evaluate options for controlling invasive mammals on islands. Ecol Appl. doi:10.1002/eap.1415
Austerlitz F, Jung-Muller B, Godelle B, Gouyon PH (1997) Evolution of coalescence times, genetic diversity and structure during colonization. Theor Popul Biol 51:148–164
Barros A, Alvarez D, Velando A (2014) Long-term reproductive impairment in a seabird after the Prestige oil spill. Biol Lett 10:20131041
Barros Á, Romero R, Munilla I, Pérez C, Velando A (2016) Behavioural plasticity in nest-site selection of a colonial seabird in response to an invasive carnivore. Biol Invasions 18:3149–3161
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. In: Laboratoire Génome, populations, interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier
Bifolchi A, Picard D, Lemaire C, Cormier JP, Pagano A (2010) Evidence of admixture between differentiated genetic pools at a regional scale in an invasive carnivore. Conserv Genet 11:1–9
Björsson TE, Heirstensson P (1991) Mink in southern Breidfjordur Bay. Wildlife Management News (Iceland), 7, 3–12. In: Macdonald D, Strachan R (eds) The mink and the water vole. Analyses for Conservation. Wildlife Conservation Research Unit and the Environment Agency, Oxford
Blackburn TM, Cassey P, Duncan RP, Evans KL, Gaston KJ (2004) Avian extinction and mammalian introductions on oceanic islands. Science 305:1955–1958
Bock DG, Caseys C, Cousens RD, Hahn MA, Heredia SM, Hübner S, Turner KG, Whitney KD, Rieseberg LH (2015) What we still don’t know about invasion genetics. Mol Ecol 24:2277–2297
Bonesi L, Macdonald DW (2004) Impact of released Eurasian otters on a population of American mink: a test using an experimental approach. Oikos 106:9–18
Bonesi L, Palazón S (2007) The American mink in Europe: status, impacts, and control. Biol Conserv 134:470–483
Bonesi L, Harrington LA, Maran T, Sidorovich VE, Macdonald DW (2006) Demography of three populations of American mink Neovison vison in Europe. Mammal Review 36:98–106
Bonesi L, Rushton SP, Macdonald DW (2007) Trapping for mink control and water vole survival: identifying key criteria using a spatially explicit individual based model. Biol Conserv 136:636–650
Borgatti SP (2002) Netdraw network visualization. Analytic Technologies, Harvard
Caswell H (2001) Matrix population models: construction, analysis, and interpretation, 2nd edn. Sinauer Associates, Sunderland
Courchamp F, Chapuis J-L, Pascal M (2003) Mammal invaders on islands: impact, control and control impact. Biol Rev 78:347–383
Courchamp F, Berec L, Gascoigne J (2008) Allee effects in ecology and conservation. Oxford University Press, New York
Craik C (1997) Long-term effects of North American Mink Mustela vision on seabirds in western Scotland. Bird Study 44:303–309
Dawson J, Oppel S, Cuthbert RJ, Holmes N, Bird JP, Butchart SH, Spatz DR, Tershy B (2015) Prioritizing islands for the eradication of invasive vertebrates in the United Kingdom overseas territories. Conserv Biol 29:143–153
Delibes M (1983) Distribution and ecology of the Iberian Carnivores: a short review. Act. XV Cong. Int. Biol. Caza, 359–378. Trujillo, España. In: Vidal T, Delibes M (Eds.) Primeros datos sobre el visón americano (Mustela vison) en el suroeste de Galicia y noroeste de Portugal. Ecología 1, 145–152
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Dunstone N (1993) The mink. T. & A.D. Poyser Ltd., London
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: Why, how and so what? Mol Ecol 19:4113–4130
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Facon B, Pointier JP, Jarne P, Sarda V, David P (2008) High genetic variance in life-history strategies within invasive populations by way of multiple introductions. Curr Biol 18:363–367
Fernández J, Toro MA (2006) A new method to estimate relatedness from molecular markers. Mol Ecol 15:1657–1667
Fitzpatrick BM, Fordyce JA, Niemiller ML, Reynolds RG (2012) What can DNA tell us about biological invasions? Biol Invasions 14:245–253
Fleming MA, Ostrander EA, Cook JA (1999) Microsatellite markers for American mink (Mustela vison) and ermine (Mustela erminea). Mol Ecol 8:1352–1354
Forsman A (2014) Effects of genotypic and phenotypic variation on establishment are important for conservation, invasion, and infection biology. Proc Natl Acad Sci 111:302–307
Fraser EJ, Macdonald DW, Oliver MK, Piertney S, Lambin X (2013) Using population genetic structure of an invasive mammal to target control efforts—an example of the American mink in Scotland. Biol Conserv 167:35–42
Fraser CI, Banks SC, Waters JM (2015) Priority effects can lead to underestimation of dispersal and invasion potential. Biol Invasions 17:1–8
Funk JL (2015) Invasive species: a global problem in need of a global solution. BioSci biv053
Gerell R (1970) Population studies on mink Mustela vison in southern Sweden. Oikos 8:83–109
Guo SW, Thompson EA (1992) A Montecarlo method for combined segregation and linkage analysis. Am J Hum Genet 51:1111–1126
Hammershøj M, Pertoldi C, Asferg T, Moller TB, Kristensen NB (2005) Danish free-ranging mink populations consist mainly of farm animals: evidence from microsatellite and stable isotope analyses. J Nat Conserv 13:267–274
Hood GM (2010) PopTools version 3.2.5. url http://www.poptools.org
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555
Kalinowski ST, Wagner AP, Taper ML (2006) ML-Relate: a computer program for maximum likelihood estimation of relatedness and relationship. Mol Ecol Notes 6:576–579
Keitt B, Campbell K, Saunders A, Clout M, Wang Y, Heinz R, Newton K, Tershy B (2011) The global islands invasive vertebrate eradication database: a tool to improve and facilitate restoration of island ecosystems. In: Veitch C, Clout M, Towns D (eds) Island invasives: eradication and management. IUCN, Gland, pp 74–77
Lawson Handley JL, Estoup A, Evans DM, Thomas CE, Lombaert E, Facon B, Roy HE (2011) Ecological genetics of invasive alien species. Biocontrol 56:409–428
Le Corre V, Kremer A (1998) Cumulative effects of founding events during colonisation on genetic diversity and differentiation in an island and stepping-stone model. J Evol Biol 11:495–512
Le Roux J, Wieczorek AM (2009) Molecular systematics and population genetics of biological invasions: towards a better understanding of invasive species management. Ann Appl Biol 154:1–17
Lecis R, Ferrando A, Ruiz-Olmo I, Manas S, Domingo-Roura X (2008) Population genetic structure and distribution of introduced American mink (Mustela vison) in Spain, based on microsatellite variation. Conserv Genet 9:1149–1161
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17:386–391
Lockwood JL, Hoopes MF, Marchetti MP (2009) Invasion Ecology. Blackwell Publishing, Oxford
Macdonald DW, Harrington LA (2003) The American mink: the triumph and tragedy of adaptation out of context. N Z J Zool 30:421–441
Manchester SJ, Bullock JM (2000) The impacts of non-native species on UK biodiversity and the effectiveness of control. J Appl Ecol 37:845–864
McDonald RA, O’Hara K, Morrish DJ (2007) Decline of invasive alien mink (Mustela vison) is concurrent with recovery of native otters (Lutra lutra). Divers Distrib 13:92–98
Medina FM, Bonnaud E, Vidal E, Nogales M (2014) Underlying impacts of invasive cats on islands: not only a question of predation. Biodivers Conserv 23:327–342
Melero Y, Robinson E, Lambin X (2015) Density-and age-dependent reproduction partially compensates culling efforts of invasive non-native American mink. Biol Invasions 17:2645–2657
Michalska-Parda A, Brzeziński M, Zalewski A, Kozakiewicz M (2009) Genetic variability of feral and ranch American mink Neovison vison in Poland. Acta Theriol 54:1–10
Moore NP, Roy SS, Helyar A (2003) Mink (Mustela vison) eradication to protect ground-nesting birds in the Western Isles, Scotland, United Kingdom. N Z J Zool 30:443–452
Nei M (1972) Genetic distance between populations. Am Nat 106:283–291
Nordström M, Högmander J, Nummelin J, Laine J, Laanetu N, Korpimäki E (2002) Variable response of waterfowl breeding populations to long-term removal of introduced American mink. Ecography 25:385–394
O’Connell M, Wright JM, Farid A (1996) Development of PCR primers for nine polymorphic American mink Mustela vison microsatellite loci. Mol Ecol 5:311–312
Paetkau D, Slade R, Burden M, Estoup A (2004) Direct, real-time estimation of migration rate using assignment methods: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Pereira P (2006) Estudio de la metodología para el control de la población de visón americano en el archipiélago de Sálvora en el Parque Nacional Islas Atlánticas de Galicia. Unpublished report
Pimentel D (ed) (2011) Biological invasions: economic and environmental costs of alien plant, animal, and microbe species. CRC Press, Boca Raton
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Ratcliffe N, Craik C, Helyar A, Roy S, Scott M (2008) Modelling the benefits of American Mink Mustela vison management options for terns in west Scotland. Ibis 150:114–121
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283
Reaser JK, Meyerson LA, Cronk Q, De Poorter M, Eldrege LG, Green E, Kairo M, Latasi P, Mack RN, Mauremootoo J, O’Dowd D, Orapa W, Sastroutomo S, Saunders A, Shine C, Thrainsson S, Vaiutu L (2007) Ecological and socioeconomic impacts of invasive alien species in island ecosystems. Environ Conserv 34:98–111
Robertson BC, Gemmell NJ (2004) Defining eradication units in pest control programmes. J Appl Ecol 41:1032–1041
Rodrigues DC, Simões L, Mullins J, Lampa S, Mendes RC, Fernandes C, Rebelo R, Santos-Reis M (2015) Tracking the expansion of the American mink (Neovison vison) range in NW Portugal. Biol Invasions 17:13–22
Rodríguez-Ramilo ST, Toro MA, Fernndez J (2009) Assessing population genetic structure via the maximisation of genetic distance. Genet Sel Evol 41:49
Rollins LA, Browning LE, Holleley CE, Savage JL, Russell AF, Griffith SC (2012) Building genetic networks using relatedness information: a novel approach for the estimation of dispersal and characterization of group structure in social animals. Mol Ecol 21:1727–1740
Roy S (2011) Strategies to improve landscape scale management of mink populations in the west coast of Scotland: lessons learned from the Uists 2001–2006. In: Veitch CR, Clout MN, Towns DR (eds) Island invasives: eradication and management. IUCN, Gland, pp 114–117
Roy SS, Chauvenet ALM, Robertson PA (2015) Removal of American mink (Neovison vison) from the Uists, Outer Hebrides, Scotland. Biol Invasions 17:2811–2820
Simberloff D (2009) The role of propagule pressure in biological invasions. Annu Rev Ecol Syst 40:81–102
Simberloff D (2011) How common are invasion-induced ecosystem impacts? Biol. Invasions 13:1255–1268
Szpiech ZA, Rosenberg NA (2011) On the size distribution of private microsatellite alleles. Theor Popul Biol 80:100–113
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24:2498–2504
Takezaki N, Nei M, Tamura K (2014) POPTREEW: web version of POPTREE for constructing population trees from allele frequency data and computing some other quantities. Mol Biol Evol 31:1622–1624
Thirstrup JP, Ruiz-Gonzalez A, Pujolar JM, Larsen PF, Jensen J, Randi E, Zalewski A, Pertoldi C (2015) Population genetic structure in farm and feral American mink (Neovison vison) inferred from RAD sequencing-generated single nucleotide polymorphisms. J Anim Sci 93:3773–3782
Tobin PC, Berec L, Liebhold AM (2011) Exploiting Allee effects for managing biological invasions. Ecol Lett 14:615–624
Veale AJ, Clout MN, Gleeson DM (2012) Genetic population assignment reveals a long-distance incursion to an island by a stoat (Mustela erminea). Biol Invasions 14:735–742
Veale AJ, Edge KA, McMurtrie P, Fewster RM, Clout MN, Gleeson DM (2013) Using genetic techniques to quantify reinvasion, survival and in situ breeding rates during control operations. Mol Ecol 22:5071–5083
Velando A, Munilla I (2011) Conservación del Cormorán moñudo en el Parque Nacional de las Islas Atlánticas. Boletín del Grupo Ibérico de Aves Marinas 35:5–14
Vidal T, Delibes M (1987) Primeros datos sobre el visón americano (Mustela vison) en el Suroeste de Galicia y Noroeste de Portugal. Ecología 1:145–152
Wagner AP, Creel S, Kalinowski ST (2006) Estimating relatedness and relationships using microsatellite loci with null alleles. Heredity 97:336–345
Wang J (2009) A new method for estimating effective population sizes from a single sample of multilocus genotypes. Mol Ecol 18:2148–2164
Wang J (2013) An improvement on the maximum likelihood reconstruction of pedigrees from marker data. Heredity 111:165–174
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Zalewski A, Piertney SB, Zalewska H, Lambin X (2009) Landscape barriers reduce gene flow in an invasive carnivore: geographical and local genetic structure of American mink in Scotland. Mol Ecol 18:1601–1615
Zalewski A, Michalska-Parda A, Bartoszewicz M, Kozakiewicz M, Brzeziński M (2010) Multiple introductions determine the genetic structure of an invasive species population: American mink Neovison vison in Poland. Biol Conserv 143:1355–1363
Zuberogoitia I, Zabala J, Martínez JA (2006) Evaluation of sign surveys and trappability of American mink: Consequences for management. Folia Zool 55:257–263
Acknowledgements
We are grateful to the staff at the Atlantic Islands of Galicia National Park and lighthouse keepers in the Island of Sálvora (Pepe, Julio and Carmen) for logistic support all over the study period. This study is based on the trapping programme performed by the National Park (Xunta de Galicia funds) with the active participation of the staff in Cíes and Sálvora islands. We are indebted to José Antonio Fernández, Director of the National Park, for his substantial support during this study. We also thank the Associate Editor and two anonymous reviewers for their helpful suggestions. Funding for this study was provided by the Spanish Ministerio de Agricultura, Alimentación y Medio Ambiente (Organismo Autónomo Parques Nacionales, 275/2011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Alberto Velando, Paloma Morán and Rafael Romero have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Velando, A., Morán, P., Romero, R. et al. Invasion and eradication of the American mink in the Atlantic Islands National Park (NW Spain): a retrospective analysis. Biol Invasions 19, 1227–1241 (2017). https://doi.org/10.1007/s10530-016-1326-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-016-1326-8