
Abstract
Investigating the origins of invasive populations provides insight into the evolutionary and anthropogenic factors underlying invasions, and can inform management decisions. Invasive species introduced for horticultural purposes often have complex origins typified by multiple introductions of species, cultivars, and genotypes, and interspecific and intraspecific hybridizations in introduced ranges. Such complex introduction histories may result in complex genetic signatures in the invaded range, making inferences about origins difficult, particularly when all putative sources cannot be sampled. In this study, we inferred the origins of the invasive French broom complex in California using 12 nuclear microsatellite markers. We characterized the genetic diversity and population structure of invasive and horticultural brooms in their invaded range in California and of Genista monspessulana in its native Mediterranean range. Overall, no significant differences in allelic richness, observed heterozygosity, inbreeding, or genetic structure were observed between the invaded and native ranges, but differences existed among populations within ranges. Bayesian STRUCTURE analysis revealed three genetic clusters in the French broom complex. Nearly all native G. monspessulana assigned highly to a single cluster. Many invasives assigned to a second cluster that contained Genista canariensis, Genista stenopetala, and ornamental sweet broom, and the remaining invasives assigned to a third cluster that also contained some G. monspessulana individuals from Sardinia and Corsica. Admixture between the second and third clusters was detected. Approximate Bayesian Computation analysis of six alternative scenarios supported the hypothesis that some invasive French broom is derived from an unsampled population branching from ornamental sweet broom. A combination of factors, including multiple introductions, escapes from cultivation, and inter-taxon hybridization, likely contribute to the invasive success of French broom in California and may have important implications for management, in particular biological control.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Invasive species pose major environmental and economic threats, but they also provide unique opportunities to study contemporary evolution (Sakai et al. 2001). In spite of considerable research focusing on the ecological and evolutionary factors that underlie successful invasions, the role of genetic diversity in promoting invasive success remains unclear. Evolutionary processes such as bottlenecks and genetic drift are expected to cause a decrease in the genetic diversity of an invasive species in its novel range (Barrett and Kohn 1991). Although this is the case for some invasions (e.g. Puillandre et al. 2008; Alexander et al. 2009), many have similar or even increased genetic diversity in invaded ranges relative to the native range (e.g. Bossdorf et al. 2005; Marrs et al. 2008). Such high genetic diversity in an invaded range might be caused by multiple introductions or a single introduction of individuals from genetically distinct source populations. Subsequent intra-specific hybridization can further increase genetic diversity in the novel range. The resulting genetic diversity may be important for the success of many invasive species because populations with low genetic variation risk inbreeding, reduced adaptive potential, and extinction (Barrett and Kohn 1991; Ellstrand and Elam 1993).
Horticulturally introduced invasive species provide ideal study systems to investigate the role of genetic diversity in plant invasions, as well as the source(s) of this diversity. These species often have complex origins resulting from multiple introductions of species, cultivars, and genotypes during the search, development, and distribution of new cultivated individuals (e.g. Okada et al. 2007; Kleist and Jasieniuk 2011). Sources of individuals might include genetically differentiated populations or taxa in the native range, different cultivars and species from breeding programs, and naturalized individuals from landscape plantings. Such complex introduction histories may profoundly affect the invasive potential of plants at any point in the invasion process (Wilson et al. 2009). The presence of pre-adapted traits may be particularly important during initial introduction and establishment (Mueller-Schaerer and Steinger 2004; Henery et al. 2010), and introducing a variety of individuals through cultivation and sale increases the chance that a pre-adapted genotype will reach a suitable habitat in the new range. Individuals from different sources can also hybridize to produce new genotypes on which selection can act (Lavergne and Molofsky 2007). Subsequent invasive spread may be promoted by repeated introductions from landscape plantings, which would increase the likelihood of individuals finding suitable sites for naturalization and reduce the probability of local population extinctions (Mack 2000). Invasive success would then be a function of propagule pressure, i.e. a composite measure of the number of individuals in an introduction event and the number of introduction events (reviewed in Lockwood et al. 2005). More recent cultivars or genotypes in landscape plantings might also contribute to invasive spread via admixture with naturalized invasive populations from an original introduction. Thus, horticultural species are typically characterized by many of the primary drivers of plant invasiveness, i.e. multiple introductions, adaptation, intra- and interspecific hybridization, and propagule pressure.
The horticulturally introduced French broom complex is highly invasive in California (Bossard 2000), with an introduction history that includes multiple ornamental sources and hybridization. In an earlier study (Kleist and Jasieniuk 2011), we found that invasive populations are comprised of (1) Genista monspessulana, (2) an unidentified species closely related to G. canariensis, G. stenopetala, and the ornamental sweet brooms, and (3) hybrids between these two groups. Individuals are monoecious, outcrossing woody legumes that are pollinated by nonnative honeybees (Parker and Haubensak 2002) and likely diploid (Cubas et al. 2001; Kang et al. 2007). Genista monspessulana is native to the Mediterranean region and the Azores islands and was first introduced into the San Francisco Bay Area of California in the mid-1800s as an ornamental plant (Bossard 2000). Genista stenopetala and G. canariensis are range-restricted endemics from the Canary Islands that may have been introduced horticulturally into California (Wojciechowski 2011), although they are not currently sold in the ornamental trade. Phylogenetically, both these species fall within the monspessulana clade (Percy and Cronk 2002), indicating a close relationship to G. monspessulana. Sweet broom, another putative contributor to invasive French broom populations in California, is currently sold as an ornamental plant throughout California although its identity is unclear, being sold under a variety of names, including Cytisus x spachianus, Teline stenopetala ssp spachiana, C. racemosus ‘Nana’, C. praecox ‘Nana’, and G. racemosa. Hybridization is likely possible between any of the French broom complex species, as G. stenopetala and G. canariensis are known to hybridize in Tenerife (Suarez Rodgriguez 1991) and the ornamental Cytisus ‘Porlock’ is thought to be a hybrid between G. monspessulana and sweet broom (AW Sheppard, CSIRO, personal communication).
In this study, we investigated the origins and population histories of French broom in California by characterizing the genetic diversity and structure of invasive and native G. monspessulana populations and the genetic signatures of G. stenopetala, G. canariensis, and ornamental plants using microsatellite data. We also performed an approximate Bayesian computation (ABC) analysis (Beaumont et al. 2002) to test alternative hypotheses concerning the contributions of G. stenopetala, G. canariensis, ornamental sweet broom, and unsampled populations to invasive French broom in California. ABC uses molecular and historical data for model-based inference of complex demographic scenarios, such as the introduction histories of invasive species that include multiple introductions, genetic admixture, population bottlenecks, and hypothetical unsampled populations (Estoup and Guillemaud 2010). Specifically, our objectives were: (1) to assess and compare the genetic diversity and population structure of native and introduced French brooms, (2) to infer the genetic origin(s) of invasive populations, and (3) to identify the most likely scenario describing the contribution of G. canariensis, G. stenopetala, ornamental sweet broom, and/or hybrids to invasive populations.
Methods
Sample collection
Individuals were sampled for leaf tissue from 29 invasive populations of the French broom complex in California, 13 populations of G. monspessulana in Europe, 14 ornamental sweet broom plants, and 12 sources of named Genista species from the monspessulana clade identified in Kleist and Jasieniuk (2011). All individuals were sampled in populations with fewer than 32 plants, whereas a stratified random sampling design (Lowe et al. 2004) was used to sample 32 plants in larger populations. For these larger populations, the area occupied by each population was divided into equally sized sections and then individuals were randomly sampled within each section for a total of 32 individuals per population. In Europe, 18–30 plants from each of five native populations were sampled for leaf tissue whereas seeds were collected from a minimum of 13 plants in eight populations, germinated, and leaf tissue sampled from the seedlings. Sampling of ornamental sweet broom included landscape plantings near invasive populations and plants from growers and garden centers. Leaf samples of Genista species from the monspessulana clade were obtained from botanical gardens, arboreta, and plant conservation programs. Following collection, leaves were either dried in silica gel or frozen at −80 °C and stored until DNA extraction.
Microsatellite marker development
DNA isolated from one individual of invasive G. monspessulana from the Auburn State Recreation Area (Asb) was used by Genetic Marker Services (Brighton, UK; www.geneticmarkerservices.com) to develop and test 12 microsatellite primer pairs (Online Resource 1) using an enriched library protocol. The primer sets were designed to amplify products ranging from 100 to 250 bp to minimize overlap ambiguities during multiplexed genotyping. Each primer pair was tested for specificity and polymorphism on high resolution agarose gels before being used for fluorescent-labeled genotyping on an ABI 3100 Genetic Analyzer (Applied Biosystems). Forward primers were labeled with 6-FAM, HEX, or NED (Applied Biosystems).
Genotyping
Total DNA was extracted from dry or frozen leaf tissue from each plant using liquid nitrogen pulverization followed by CTAB extraction (Doyle and Doyle 1987). All individuals were genotyped at 12 microsatellite loci in five multiplexed reactions (Multiplex A: uc24, uc26, uc29; Multiplex B: uc34, uc38; Multiplex C: uc36, uc37, uc39; Multiplex D: uc5, uc6; Multiplex E: uc3, uc7), each in a total volume of 12 μL. Each reaction contained 20 ng of template DNA, 1x PCR buffer (Qiagen) containing 1.5 mM MgCl2, 0.1–0.3 mM of each primer depending on the individual locus (Online Resource 1), 0.2 mM of each dNTP, and 1 unit of Taq polymerase. An annealing temperature of 56 °C was used for Multiplexes A, C, and D, 54 °C was used for Multiplex B, and 59 °C was used for Multiplex E. Amplifications were carried out in an MJ Research PTC-200 thermal cycler (Bio-Rad Laboratories) using the following conditions: an initial denaturation at 94 °C for 5 min, annealing at 56 °C, 52 °C, or 59 °C for 1 min, extension at 72 °C for 1 min, and a final extension at 72 °C for 30 min. PCR products were genotyped on an ABI 3100 Genetic Analyzer. Fragments were sized using GeneMapper software version 3.7 (Applied Biosystems) with GeneScan 400HD ROX (Applied Biosystems) size standard for reference. Approximately 5 % of samples were amplified at least twice with all multiplexes in separate reactions to assess repeatability. In addition, individuals with rare or unusual alleles were re-amplified to confirm their genotypes.
Data analysis
Genetic diversity of invasive, native, and ornamental brooms
To quantify the genetic diversity of the sampled invasive, native, and ornamental individuals at each of the 12 microsatellite loci, we determined the total number of distinct alleles (T A) detected at each locus for each group. The program MICRO-CHECKER (Van Oosterhout et al. 2004) was used to check for null alleles. Wright’s inbreeding coefficient (F IS) was estimated for each invasive and native population and averaged over all invasive and native populations for each locus using the software FSTAT 2.9.3 (Goudet 2001). Wright’s fixation index (F ST) was estimated over the 29 invasive populations and the 13 native populations at each locus. Lastly, pairwise F ST was estimated between French broom populations after sequential Bonferroni correction, also using FSTAT.
To estimate genetic diversity within populations, we calculated allelic richness (A), expected heterozygosity (H E), observed heterozygosity (H O), and F IS for each invasive and native population. All calculations were performed using GenAlEx version 6.41 (Peakall and Smouse 2006), with the exception of allelic richness, which was performed using FSTAT version 2.9.3 (Goudet 2001). For both exhaustively sampled invasive populations (n ≤ 32) and larger populations from which only 32 individuals were sampled, allelic richness was calculated as the mean number of alleles detected in all individuals sampled in the population. We did not correct for differences in population size using a rarefaction procedure because the same number of individuals were sampled in populations with more than 32 individuals. Departure from Hardy–Weinberg equilibrium (HWE) of each population at each locus, and linkage disequilibrium (LD) between each pair of polymorphic loci for each population, were tested using GENEPOP version 4.1.4 (Rousset 2008) with a Markov chain approximation of exact tests and likelihood-ratio tests, respectively.
The distribution of genetic variation between invaded and native regions was examined with statistical comparisons of genetic diversity indices. The allelic richness (A), observed heterozygosity (H O), inbreeding coefficient (F IS), and fixation index (F ST) for the invaded and native regions were calculated using FSTAT and compared statistically using 1,000 permutations with the Bonferroni procedure (Rice 1989).
Population structure
To infer population structure, assign invasive, native, and ornamental individuals to populations, and test for admixture among taxa and populations, we performed Bayesian clustering analyses implemented in the program STRUCTURE version 2.3.3 (Pritchard et al. 2000). STRUCTURE clusters individuals into K distinct populations by minimizing Hardy–Weinberg and linkage disequilibrium within populations. All sampled individuals are assigned probabilistically to clusters or jointly to several clusters if their genotypes are admixed. STRUCTURE was run using the ‘admixture model’ and correlated allele frequencies with 100,000 MCMC repetitions and a 50,000 burn-in period. The number of populations or clusters (K) was set from 1 to 10, with each K replicated independently 15 times for the total data set and again 15 times each for the native, ornamental, and invasive data subsets. To identify the most likely K, we used the absolute values of the second order rate of change of the likelihood distribution divided by the standard deviation of the likelihoods (∆K), following Evanno et al. (2005). For the identified K clusters for each dataset, an individual’s assignment coefficient (q) to each genetic cluster was averaged across all 15 runs using the CLUMPP software (Jakobsson and Rosenberg 2007) and visualized using DISTRUCT 1.1 (Rosenberg 2003).
Origin of distinct subset of invasive populations
A previous study (Kleist and Jasieniuk 2011) identified a distinct subset of invasive French broom populations in California with close phylogenetic relatedness to G. canariensis, G. stenopetala, and ornamental sweet broom (also see STRUCTURE results of current study). However, it was not possible to determine whether G. canariensis, G. stenopetala, ornamental sweet broom, or a hybrid between these contributed to the invasive populations. To investigate the origins of this population subset, we performed approximate Bayesian computation (ABC) analysis. The ABC analysis was conducted using DIYABC (Cornuet et al. 2010) and evaluated six possible scenarios of population origins describing how G. canariensis, G. stenopetala, and ornamental sweet broom may have contributed to the subset of invasive populations: (1) Genista species in the monspessulana clade are the direct source of invasives, (2) sweet broom is the direct source of invasives, (3) a hybrid between Genista species in the monspessulana clade and sweet broom is the direct source of invasives, (4) an unsampled population diverging from ornamental sweet broom is the source of invasive plants, (5) an unsampled population diverging from Genista species in the monspessulana clade is the source of invasives, and (6) a hybrid between an unsampled population diverging from Genista species in the monspessulana clade and sweet broom is the source of invasives. Because our sampling may represent only a subset of possible source populations, three scenarios also included unsampled source populations.
Model parameters were defined based on historical knowledge of the introductions. Uniform prior values for t1 were bounded by 1 and 150 and values for the bottleneck period were bounded by 1 and 10 generations. In cases where little information was available, such as the likely effective population sizes of each species in each location and the time of split between sweet broom and Genista species, broad priors were used. A uniform prior bounded by 10 and 100,000 was used for the effective population size of Genista species and by 10 and 10,000 for the effective population size of sweet broom and unsampled populations. The generalized stepwise model (GSM), which assumes increases or decreases in one or more microsatellite repeat units, was used as the mutational model for microsatellites (Fu and Chakraborty 1998). We used the means of Nei’s gene diversity (H T ) and Garza–Williamson’s M as the summary statistics for each genetic group following Cornuet et al. (2008). Mean genic diversity, F ST, and the mean individual assignment likelihood (Pascual et al. 2007) were used as summary statistics for each pair of genetic groups. Three million simulations were run, producing 500,000 simulated data sets for each scenario. Models were compared by estimating their posterior probabilities using both a direct estimate and logistic regression on the 10 % closest simulated points, as described in Cornuet et al. (2010).
Results
Genetic diversity
Within loci
We detected 231 distinct alleles in the 1,060 individuals (703 invasive French broom, 330 native G. monspessulana, 14 ornamental sweet broom, and 13 other Genista species from the monspessulana clade) that were sampled (see Tables 1 and 2) and genotyped at the 12 microsatellite loci. The total number of alleles in invasive French broom was 156, ranging from 5 to 27 per locus (Online Resource 2). The total number of alleles in G. monspessulana from its native Mediterranean range was 139, ranging from 4 to 38 per locus. Of the 231 alleles detected over all the genotyped plants, 31 alleles were unique to invasive French broom and 38 alleles were unique to native G. monspessulana. No private alleles were detected in ornamental sweet broom, and all alleles found in these ornamentals were also found in invasive French broom populations.
The inbreeding coefficient F IS varied widely among loci, ranging from −0.054 to 0.317 in invasives and from −0.033 to 0.387 in natives (Online Resource 2). The most extreme F IS values were not found at the same loci in the invaded and native ranges. High F IS values could be caused by null alleles, which can appear due to technical problems with amplification and scoring or because of mutations in the sequence used for primer design. However, evidence for null alleles was not detected in our dataset. Global tests for heterozygote deficiency revealed significant deficiencies at one locus (uc26) in the invaded range and one locus (uc 3) in the native range. F ST indicated significant differentiation among populations at all loci, with higher values for invasives than natives.
Within populations
Within invasive French broom, levels of diversity as measured by allelic richness (A), expected heterozygosity (H E ), and observed heterozygosity (H O ) varied widely among populations (Table 2). Allelic richness ranged from 1.6 to 6.8, mean expected heterozygosity ranged from 0.07 to 0.71, and mean observed heterozygosity ranged from 0.03 to 0.68 among populations. Linkage disequilibrium (LD) was detected in only four of the possible comparisons of pairs of loci within populations. LD between the same pairs of loci was not found in populations from the native range, which is consistent with independently segregating loci. A significant deviation from Hardy–Weinberg equilibrium was observed in 53 out of 348 population-by-locus test combinations, caused by a deficiency in heterozygotes globally. F IS within populations reflected this deficiency and significant positive values were found in 26 out of 29 populations. The most inland populations (JU, WH, and SCB) had the lowest allelic richness estimates and highest F IS values.
Within native range G. monspessulana, levels of heterozygosity and allelic richness were more homogeneous across populations than those observed in invasive French broom (Table 2). Allelic richness ranged from 2.4 to 5.5, mean expected heterozygosity ranged from 0.25 to 0.60, and mean observed heterozygosity ranged from 0.25 to 0.53. Deviations from HWE were observed in 17 out of 144 population-by-locus test combinations and a significant heterozygote deficiency was observed in only two populations (LD and SF). Overall, F IS values were lower than those estimated in invasive French broom populations.
Between regions
We did not observe a significant difference in allelic richness, observed heterozygosity, F IS , or F ST values between the invaded and native ranges (Table 3).
Population structure
Pairwise F ST values between invasive populations ranged from 0.039 to 0.861 (Online Resource 3). The lowest pairwise F ST values were found between populations in close proximity in the San Francisco Bay area. In comparison, pairwise F ST values between native populations ranged from 0.133 to 0.549 (Online Resource 3).
To identify the origins of invasive French broom in California, Bayesian clustering was first performed with all sampled individuals. Consistent results were found with the program STRUCTURE over the 15 runs at each K. The statistic ∆K (Evanno et al. 2005) indicated that three clusters best explained the uppermost hierarchical level of genetic structuring across individuals of native, invasive, and ornamental brooms (Online Resource 4). The vast majority of G. monspessulana individuals from the native range assigned highly (q > 0.9) to Cluster 1 (Fig. 1). All individuals of ornamental sweet broom, 12 out of 13 individuals of Genista species other than G. monspessulana, and 36 % of individuals of invasive French broom from California assigned highly (q > 0.9) to Cluster 2. Of the remaining invasive individuals, 78 % assigned highly to Cluster 3, while 22 % were admixed with genomes originating in both Clusters 2 and 3 (Fig. 1). The results agree with a recent molecular phylogenetic study that found that many invasive French broom populations contained hybrids (Kleist and Jasieniuk 2011). All hybrid populations identified by Kleist and Jasieniuk (2011) also contained admixed individuals in the STRUCTURE analysis.

Assignment of ornamental sweet broom, Genista species, native, and invasive individuals to the three clusters identified by STRUCTURE analysis at the highest hierarchical level of genetic structure. Each vertical bar represents an individual and the proportion of its genome that assigns to the two clusters (medium grey = Cluster 1; white = Cluster 2; dark grey = Cluster 3). Individuals are grouped by sampling location in the native and invaded ranges. Population IDs correspond to those in Table 2
Subsequent STRUCTURE analyses to detect genetic substructuring (sensu Coulon et al. 2008) within Clusters 1, 2, and 3 revealed subgroups within each cluster. Two subgroups were identified within Cluster 1 (Online Resource 4; Fig. 2). G. monspessulana individuals from the native range assigned to the two subgroups according to their geographical origins with individuals from Sardinia, Corsica, and southeastern France assigning highly to one subgroup and individuals from southwestern France and Spain assigning highly to a second subgroup (Fig. 2a). A population from Ganges, France (GG), which is geographically intermediate, consisted of individuals that assigned to both subgroups within Cluster 1. Two subgroups were also identified within Cluster 2 (Online Resource 4; Fig. 3A). Individuals of ornamental sweet broom, Genista species other than G. monspessulana, and three invasive populations (LC, PLA, and SBB) assigned highly to one of the two Cluster 2 subgroups (Fig. 3a) but the vast majority of invasive individuals assigning to Cluster 2 assigned highly to the second subgroup (Fig. 3a). Interestingly, individuals from population SBA consisted almost entirely of admixed genotypes. Finally, eight genetic subgroups were identified within Cluster 3 (Online Resource 4; Fig. 3B). Populations with individuals assigning to the eight subgroups were spread across California (Fig. 3c).

Genetic structure of Genista monspessulana sampled in the native range. a Map of Europe showing the geographical distribution of the native populations sampled and the proportion of assignment of each sampled population to the two genetic subgroups identified within Cluster 1 (Fig. 1) by STRUCTURE analysis. b Assignment of native individuals to each of the two genetic subgroups (K = 2) within Cluster 1 identified by STRUCTURE analysis. Each vertical line represents an individual and each color represents a subgroup. Individuals are grouped by the populations sampled (Table 2)

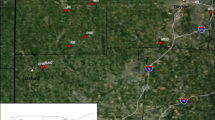
Genetic structure of individuals of invasive French broom, ornamental sweet broom, and Genista species in California. a Assignment of ornamental sweet broom, Genista species, and the invasive French broom individuals to the two (K = 2) genetic subgroups within Cluster 2 (Fig. 1) as identified by STRUCTURE analysis. Each vertical line represents an individual and each color represents a subgroup. Individuals are grouped by the populations sampled (Table 2). b Assignment of invasive French broom individuals to eight (K = 8) genetic subgroups within Cluster 3 (Fig. 1) as identified by STRUCTURE analysis. c Map of California indicating the geographical distribution of the sampled invasive populations and the proportion of assignment of each sampled population to the clusters and subgroups identified by STRUCTURE. Three populations (Ca, Sca and Val2) are not included because most individuals were admixed
Approximate Bayesian computation to infer origins of invasives
ABC analyses were used to test six competing hypotheses or scenarios (Fig. 4) for the origins of invasive individuals that assigned highly to Cluster 2 from STRUCTURE analysis. Ornamental plants currently being sold by the horticultural trade assigned to this cluster thus it was necessary to determine the origins of these horticultural types in order to suggest their removal from the marketplace. An unsampled source population was included in three of the scenarios due to the possibility that some ornamental genotypes are no longer available commercially. Scenario 4 was found to have the highest posterior probability using both the direct estimate and logistic regression approaches (Fig. 5). Because logistic regression is considered to discriminate between scenarios better than the direct estimate (Cornuet et al. 2008), only posterior probabilities from logistic regression are described here. Scenarios 1, 2, 3, and 5 were clearly rejected with posterior probabilities <0.01. Scenario 6 was also rejected with a posterior probability of <0.09. The most highly supported scenario was Scenario 4, with a very high posterior probability of 0.8987. Scenario 4 hypothesized that Genista species and ornamental sweet broom diverged in the past and then an unsampled population diverged from ornamental sweet broom. Invasive French broom then diverged from this unsampled population.

Graphical representation of six alternative hypotheses or scenarios for the origin of invasive French broom individuals assigning to Cluster 2 (Fig. 1). Individuals of Genista species (Pop 1) are represented with a dashed line, sweet broom individuals (Pop 2) are colored black, invasive French broom individuals (Pop 3) are colored dark blue, a population bottleneck is colored light teal, and unsampled population(s) are colored orange. All six scenarios assume that at the present time (0 years), there are three genetic groups, and that these diverged from a single population in the past (t3). Historical and demographic parameters were the same for all scenarios. The time scale is shown on the right of each graphic

Results of the ABC analysis showing the probabilities of each of the six scenarios for the origin of invasive French broom depicted in Fig. 4. Scenario probabilities were determined using logistic regression. Yellow line: Scenario 4; teal line: Scenario 6; dark blue line: Scenario 3; deep pink line: Scenario 5; red line: Scenario 1; green line: Scenario 2. Scenarios 1, 2, 3, 5, and 6 had the same probability, but only the deep pink line of Scenario 5 is easily visible
Discussion
The introduction history of French broom in California is complex, involving multiple closely-related species that have been sold as ornamental plants throughout the state, likely under incorrect species names. These horticultural introductions have led to an invasive complex that is genetically very different from native G. monspessulana populations. Despite the observed genetic differentiation from native populations, invasive broom populations in California retain unique and reasonably high genetic diversity, which may be the result of numerous processes, including multiple introductions (e.g. Thompson et al. 2012), adaptation to variable habitats (e.g. Lavergne and Molofsky 2007), and intra- and interspecific hybridization (e.g. Culley and Hardiman 2009).
Interestingly, two genetically distinct subgroups were found within the native Mediterranean region of G. monspessulana, and may be a response to historical glaciations. The subgroups were separated by a population containing individuals from both subgroups, and although it is possible that this population structure is due to local adaptation, it seems unlikely given the similar habitats of individuals from both subgroups in southern France. Rather, this genetic clustering may suggest that there has been a long-term barrier to gene flow between populations in Spain and southwestern France on the one hand, and populations from southeastern France, Corsica, and Sardinia on the other. The two subgroups may be descended from two separate refugia established during the Pleistocene or later glacial maxima. Similar patterns have been seen in other groups (e.g. Schonswetter et al. 2002; Breton et al. 2006; Boratyński et al. 2009), and our sampling included populations at, or adjacent to, seven putative refugia within the Mediterranean region (Médail and Diadema 2009).
The exact native source in the Mediterranean region of invasive French broom in California is not clear. Given the strong population structure in our sampled native range, and that invasive individuals were not found to assign to the same cluster as individuals from Spain and southwestern France, we did not find evidence that individuals from these areas contributed to invasive populations in California. It is possible, however, that individuals from Corsica and Sardinia may be one source of invasive French broom. A small number of individuals from Corsica and Sardinia were admixed with genomes assigning to both the native G. monspessulana cluster (Cluster 1) and an invasive French broom cluster (Cluster 3), suggesting that individuals from this area may have contributed to the French broom invasion in California. If this is the case, bottlenecks associated with founder events and strong drift since introduction into the invaded range could have caused the observed differentiation between invasive and native populations.
Our results revealed high genetic heterogeneity among invasive populations, which may be a signature of G. monspessulana’s original introductions into California by the horticultural industry. Eight distinct genetic subgroups were identified in the invaded range, suggesting multiple horticultural introductions as G. monspessulana was sold throughout California before it was declared a noxious weed and taken off the market. No clear pattern of geographical clustering of genetic groups was evident from our results. Since many escapes from cultivation may have occurred throughout the state, the sources of some of these horticultural introductions may not have been sampled in this study. In the native range, our sampling focused on areas where G. monspessulana is abundant (AW Sheppard, CSIRO, personal communication), but additional small and scattered populations can be found surrounding the Mediterranean basin (Tutin et al. 1968) and populations in Italy, Greece, Turkey, and North Africa could have been sources of novel plant material for the horticultural industry. Such multiple horticultural introductions can result in similar amounts of genetic diversity in introduced and native populations (Novak 2007), as observed here. Multiple introductions are a common feature of invasions (Bossdorf et al. 2005; Novak and Mack 2005) that can lead to novel genetic combinations and increase variation at adaptive loci.
Hybridization is also thought to play a role in stimulating invasiveness (Ellstrand and Schierenbeck 2000; Rieseberg et al. 2007), as it can provide immediate advantages for a colonizing species through heterosis (Blum et al. 2010), and subsequent recombination and segregation can create new genotypes on which selection can act. These recombinant genotypes may provide the means for rapid adaptation to new abiotic and biotic conditions (Lavergne and Molofsky 2007), increasing invasive potential. Currently marketed ornamental brooms likely play a role in the California French broom invasion, both directly and through interspecific hybridization. In a previous phylogenetic study, we found that some individuals identified as invasive French broom were actually ornamental sweet broom, Genista species, or hybrids with naturalized French broom individuals (Kleist and Jasieniuk 2011). Although population samples were not identical for this and the earlier study, all populations that clustered with G. canariensis, G. stenopetala, and ornamental sweet broom in this study were also found in the ornamental sweet broom group in the previous phylogenetic study. Further, many invasive populations contained individuals that were admixed between ornamental and invasive clusters, showing that interspecific hybridization is relatively common in the invasive complex.
Due to the relatively small number of sweet broom individuals included in this study, it is difficult to determine whether the unsampled progenitor of the invasives clustering with G. canariensis, G. stenopetala, and ornamental sweet broom is currently sold but not collected, or whether it was sold in the past and no longer available. The substructure within the cluster containing a portion of the invasive individuals, G. canariensis, G. stenopetala, and ornamental sweet broom suggests that some invasive populations are very closely related to sweet broom, while other populations are more closely related to each other than to sweet broom. Three populations containing a large number of individuals that cluster with sweet broom are in close geographic proximity to each other, and it is possible that escape from cultivation occurred once in this group and was followed by invasive spread. The other invasive individuals in this cluster containing a portion of the invasives, G. canariensis, G. stenopetala, and ornamental sweet broom are randomly distributed, suggesting that escape from cultivation occurred recurrently within regions.
Knowledge of the origins of invasive populations is important for management efforts. Multiple horticultural species and groups are likely to have contributed to invasive French broom in California. This includes a lineage closely related to a currently sold ornamental plant, sweet broom, which contributes to some invasive French broom populations directly and via hybridization. In addition, several populations contain intra- or inter-specific hybrids descending from separate introductions, which may have led to increased success in the invaded range. Although we cannot say with certainty whether a currently marketed, or previously marketed, sweet broom contributes to invasive populations, we suggest that the sale of sweet broom should be restricted in California. There is no evidence that these plants are sterile, and it is reasonable to assume that they have the ability to become invasive.
In addition to providing information suggesting means to prevent the further spread of invasive French broom by removing sweet broom from the market in California, this study also has important implications for designing biological control programs for invasive French broom. The effectiveness of biological control agents for managing invasive plants is influenced by both the genetic diversity and the origins of invasive populations (Mueller-Schaerer and Schaffner 2008). Multiple origins, genetic variation and hybridization within invasive populations tends to limit the success of biological control programs (Burdon and Marshall 1981; Roderick and Navajas 2003), as these populations are unlikely to have host-specific and damaging natural enemies in their native range. Our study therefore suggests that biological control may have limited success against invasive French broom in California.
References
Alexander JM, Poll M, Dietz H, Edwards PJ (2009) Contrasting patterns of genetic variation and structure in plant invasions of mountains. Divers Distrib 15:502–512
Barrett SCH, Kohn JR (1991) Genetic and evolutionary consequences of small population size in plants: implications for conservation. In: Falk DA, Holsinger KE (eds) Genetics and conservation of rare plants. Oxford University Press, New York, pp 3–30
Beaumont MA, Zhang W, Balding DJ (2002) Approximate Bayesian computation in population genetics. Genetics 162:2025–2035
Blum MJ, Walters DM, Burkhead NM, Freeman BJ, Porter BA (2010) Reproductive isolation and the expansion of an invasive hybrid swarm. Biol Invasions 12:2825–2836
Boratyński A, Lewandowski A, Boratyńska K, Montserrat JM, Romo A (2009) High level of genetic differentiation of Juniperus phoenicea (Cupressaceae) in the Mediterranean region: geographic implications. Plant Syst Evol 277:163–172
Bossard CC (2000) Genista monspessulana (L.) L. Johnson. In: Bossard CC, Randall JM, Hoshovsky MC (eds) Invasive plants of California’s wildlands. UC Press, Berkeley, pp 203–208
Bossdorf O, Auge H, Lafuma L, Rogers WE, Siemann E, Prati D (2005) Phenotypic and genetic differentiation between native and introduced plant populations. Oecologica 144:1–11
Breton C, Tersac M, Bervillé A (2006) Genetic diversity and gene flow between the wild olive (oleaster, Olea europaea L.) and the olive: several Plio-Pleistocene refuge zones in the Mediterranean basin suggested by simple sequence repeats analysis. J Biogeogr 33:1916–1928
Burdon JJ, Marshall DR (1981) Biological control and the reproductive mode of weeds. J Appl Ecol 18:649–658
Cornuet JM, Santos F, Beaumont MA, Robert CP, Marin J-M, Balding DJ, Guillemaud T, Estoup A (2008) Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24:2713–2719
Cornuet JM, Ravigne V, Estoup A (2010) Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinformatics 11:401
Coulon A, Fitzpatrick JW, Bowman R, Stith BM, Makarewich CA, Stenzler LM, Lovette IJ (2008) Congruent population structure inferred from dispersal behaviour and intensive genetic surveys of the threatened Florida scrub-jay (Aphelocoma coerulescens). Mol Ecol 17:1685–1701
Cubas P, Tahiri H, Pardo C (2001) Karyological and taxonomic notes on Cytisus Desf. Sect. Spartopsis Dumort. and Sect. Alburnoides DC. (Genisteae, Leguminosae) from the Iberian Peninsula and Morocco. Bot J Linn Soc 135:43–50
Culley TM, Hardiman NA (2009) The role of intraspecific hybridization in the evolution of invasiveness: a case study of the ornamental pear tree Pyrus calleryana. Biol Invasions 11:1107–1119
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure from small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size—implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Ellstrand NC, Schierenbeck KA (2000) Hybridization as a stimulus for the evolution of invasiveness in plants? P Natl Acad Sci USA 97:7043–7050
Estoup A, Guillemaud T (2010) Reconstructing routes of invasion using genetic data: why, how and so what? Mol Ecol 19:4113–4130
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fu YX, Chakraborty R (1998) Simultaneous estimation of all the parameters of a stepwise mutation model. Genetics 150:487–497
Goudet J (2001) FSTAT, a program to estimate and test gene diversity and fixation indices (version 2.9.3), http://www2.unil.ch/popgen/softwares/fstat.htm
Henery ML, Bowman G, Mráz P, Treier UA, Gex-Fabry E, Schaffner U, Muller-Scharer H (2010) Evidence for a combination of pre-adapted traits and rapid adaptive change in the invasive plant Centaurea stoebe. J Ecol 98:800–813
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Kang M, Buckley YM, Lowe AJ (2007) Testing the role of genetic factors across multiple independent invasions of the shrub Scotch broom (Cytisus scoparius). Mol Ecol 16:4662–4673
Kleist A, Jasieniuk M (2011) A molecular phylogenetic analysis of invasive and ornamental brooms and their relationships within the Genistoid legumes. Mol Phylogenet Evol 61:970–977
Lavergne S, Molofsky J (2007) Increased genetic variation and evolutionary potential drive the success of an invasive grass. P Natl Acad Sci USA 104:3883–3888
Lockwood JL, Cassey P, Blackburn T (2005) The role of propagule pressure in explaining species invasions. Trends Ecol Evol 20:223–228
Lowe A, Harris S, Ashton P (2004) Markers and sampling in ecological genetics. In: Lowe A, Harris S, Ashton P (eds) Ecological genetics: design, analysis, and application. Blackwell, London, pp 6–45
Mack RN (2000) Cultivation fosters plant naturalization by reducing environmental stochasticity. Biol Invasions 2:111–122
Marrs RA, Sforza R, Hufbauer RA (2008) When invasion increases population genetic structure: a study with Centaurea diffusa. Biol Invasions 10:561–571
Médail F, Diadema K (2009) Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr 36:1333–1345
Mueller-Schaerer H, Steinger T (2004) Classical biological control: exploiting enemy escape to manage plant invasions. Biol Invasions 10:859–874
Mueller-Schaerer H, Schaffner U (2008) Classical biological control: exploiting enemy escape to manage plant invasions. Biol Invasions 10:859–874
Novak SJ (2007) The role of evolution in the invasion process. P Natl Acad Sci USA 104:3671–3672
Novak SJ, Mack RN (2005) Genetic bottlenecks in alien plant species: influence of mating systems and introduction dynamics. In: Sax DF, Stachowicz JJ, Gaines SD (eds) Species invasions: insights into ecology, evolution, and biogeography. Sinauer Associates, Sunderland, MA, pp 201–228
Okada M, Ahmad R, Jasieniuk M (2007) Microsatellite variation points to local landscape plantings as sources of invasive pampas grass (Cortaderia selloana) in California. Mol Ecol 16:4956–4971
Parker IM, Haubensak KA (2002) Comparative pollinator limitation of two non-native shrubs: do mutualisms influence invasions? Oecologia 130:250–258
Pascual M, Chapuis MP, Mestres F, Balanyà J, Huey RB, Gilchrist GW, Serra L, Estoup A (2007) Introduction history of Drosophila subobscura in the New World: a microsatellite based survey using ABC methods. Mol Ecol 16:3069–3083
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Percy DM, Cronk QCB (2002) Different fates of island brooms: contrasting evolution in Adenocarpus, Genista, and Teline (Genisteae, Fabaceae) in the Canary Islands and Madeira. Am J Bot 89:854–864
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puillandre N, Dupas S, Dangles O, Zeddam JL, Capdevielle-Dulac C, Barbin K, Torres-Leguizamon M, Silvain JF (2008) Genetic bottleneck in invasive species: the potato tuber moth adds to the list. Biol Invasions 10:319–333
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rieseberg LH, Kim SC, Randell RA, Whitney KD, Gross BL, Lexer C, Clay K (2007) Hybridization and the colonization of novel habitats by annual sunflowers. Genetica 129:149–165
Roderick GK, Navajas M (2003) Genes in new environments: genetics and evolution in biological control. Nat Rev Genet 4:889–899
Rosenberg NA (2003) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Sakai AK, Allendorf FW, Holt JS, Lodge DM, Molofsky J, With KA, Baughman S et al (2001) The population biology of invasive species. Annu Rev Ecol Syst 32:305–332
Schonswetter P, Tribsch A, Barfuss M, Niklfeld H (2002) Several Pleistocene refugia detected in the high alpine plant Phyteuma globulariifolium Sternb. & Hoppe (Campanulaceae) in the European Alps. Mol Ecol 11:2637–2647
Suarez Rodgriguez C (1991) Estudio de los relictos actuales del monte verde en Gran Canaria. Cabildo Insular de Gran Canaria, Las Palmas de Gran Canaria
Thompson GD, Bellstedt DU, Byrne M, Millar MA, Richardson DM, Wilson JRU, Le Roux JJ (2012) Cultivation shapes genetic novelty in a globally important invader. Mol Ecol 21:3187–3199
Tutin TG, Heywood VH, Burges NA, Moore DM, Valentine DH, Walters SM, Webb DA (1968) Flora Europaea Vol 2 Rosaceae to Umbelliferae. Cambridge University Press, UK
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wilson JRU, Dormontt EE, Prentis PJ, Lowe AJ, Richardson DM (2009) Something in the way you move: dispersal pathways affect invasion success. Trends Ecol Evol 24:136–144
Wojciechowski MF (2011) Genista. In: Baldwin BG, Goldman DH, Keil DJ, Patterson RW, Rosatti TJ (eds) The Jepson Manual: vascular plants of California, 2nd edn. UC Press, Berkeley, pp 754–756
Acknowledgments
Annabelle Kleist was supported by a Department of Plant Sciences Graduate Student Research award. Financial support for the research from the United States Department of Agriculture, Henry A. Jastro Research Scholarships, and the California Weed Science Society is gratefully acknowledged. Trammie Bui and Yuan Qin provided lab assistance. We thank Carla D’Antonio, UC Botanical Garden, Los Angeles County Arboretum and Botanic Garden, Desert Legume Program, Ökologisch-Botanischer Garten der Universität Bayreuth, Monrovia Nursery, RZ Nursery, Walter Andersen Nursery, and Merriments Gardens for plant specimens. We also thank Joseph DiTomaso for introducing us to the study system, and Libby Karn, Miki Okada, Dan Potter, and Kevin Rice for useful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kleist, A., Herrera-Reddy, A.M., Sforza, R. et al. Inferring the complex origins of horticultural invasives: French broom in California. Biol Invasions 16, 887–901 (2014). https://doi.org/10.1007/s10530-013-0546-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-013-0546-4