
Abstract
Invasiveness might depend on the ability of genetically diverse populations of exotic species to adapt to novel environments, which suggests a paradox since exotic species are expected to lose genetic diversity when introduced. The apparent need for genetic diversity is particularly important for exotic species that lack bi-parental reproduction and genetic recombination. We used genetic marker studies to determine the genotypic diversity of invasive US populations of the clonal New Zealand mudsnail (Potamopyrgus antipodarum). We report here on a three-pronged survey of allozyme, microsatellite DNA, and mitochondrial DNA genetic markers of invasive populations with a focus on the western US. Combining the three types of genetic markers, we discovered four distinct genotypes of P. antipodarum. These results show that only one genotype (US 1) occupied the vast majority of the western US range, and a second occurred in the Great Lakes in the eastern US (US 2). Two other genotypes occurred in the western US (US 1a and US 3), but were restricted to populations near the presumed source of invasion in the middle Snake River, ID. These results suggest that P. antipodarum spread across a broad geographic range in the western US from a single introduced source population, and that the populations are comprised of a single clonal lineage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many exotic species expand their range sometimes as a result of evolved phenotypic adaptation associated with niche evolution (Huey et al. 2000; reviewed in Lee 2002; Lee and Gelembiuk 2008). Genetic diversity is required for evolutionary adaptation, but a reduction of genetic diversity in invasive populations compared to their native range populations is expected and often observed (Amsellam et al. 2000; DeWalt and Hamrick 2004; Grapputo et al. 2005; Bossdorf et al. 2005, Dlugosch and Parker 2008). On the other hand, a number of studies have shown that genetic diversity can be higher among invasive populations compared to native populations due to introductions from multiple sources via strong propagule pressure (Lockwood et al. 2005) and genetic mixing (Kolbe et al. 2004; Lavergne and Molofsky 2007; Facon et al. 2008; reviewed in Roman and Darling 2007).
The importance of repeated introductions and genetic diversity is amplified for invasive species that are incapable of bi-parental sexual reproduction (e.g., parthenogenetic or selfing species). Although these species benefit from reproductive assurance, they lack the capacity to generate novel genetic variation due to the absence of bi-parental reproduction and genetic recombination. Hence, a reduction in genetic diversity in invasive species, and particularly in those lacking bi-parental reproduction, presents a paradox if evolvability is important to invasion success (Roman and Darling 2007).
Parthenogenetic (or clonal) populations of a New Zealand freshwater mudsnail (Potamopyrgus antipodarum) have invaded Europe, Australia, and the US (Ponder 1988; Wallace 1992; Zaranko et al. 1997; Dybdahl and Kane 2005). These snails undergo both sexual and clonal reproduction in their native range, and the genotypic diversity of clonal populations in their native range is very high (Dybdahl and Lively 1995). Further detailed studies in the native range showed that a wide range of niches are occupied because of a broad diversity of ecologically distinct multilocus genotypes (MLGs) (Fox et al. 1996; Jokela et al. 1999; Jokela et al. 2003). Taken together, these studies suggest that expansion across a broad range of niches might be most likely if sufficient genotypic diversity was present in introduced P. antipodarum populations. European populations, which were founded in the late 1800′s, are comprised of several clonal lineages as measured by phenotypic and molecular marker variation (Ponder 1988; Hauser et al. 1992; Weetman et al. 2002). However, less is known about the genotypic diversity of US and Australian populations.
In the US, P. antipodarum has dispersed rapidly and widely in the western US and the Great Lakes over the past 25 years, with the first populations detected in the Snake River, ID (Proctor et al. 2007). Ecological studies suggest that P. antipodarum is highly invasive (Hall et al. 2003; Kerans et al. 2005; Hall et al. 2006). Preliminary surveys of US populations of P. antipodarum using allozyme markers recovered only two different multilocus genotypes, suggesting a lack of genotypic diversity. These two genotypes were correlated with distinct geographic ranges; one of these genotypes occurred in several populations in the western US (Dybdahl and Kane 2005), and the second in populations from Lake Ontario (Proctor et al. 2007).
Here, our goal was to obtain a more detailed picture of genotypic diversity and assess the potential for niche evolution in western US populations of P. antipodarum. In this paper, we describe the results of a thorough and systematic survey using allozymes, mitochondrial DNA sequences (mtDNA), and nuclear microsatellite DNA loci (msDNA), focusing on the western US. We included rapidly evolving msDNA loci because they can detect lineages that have arisen recently, and hence reveal the presence of genotypic diversity that is not detectable by slowly evolving allozymes or mtDNA.
Methods
Sample collection
We collected samples from populations across the western US (see Table 1; Fig. 1). Sampling was designed to cover presumed regional centers of invasion in the western US, based on the historical sequence of first reports and historical pattern of dispersal. In a few cases, samples were obtained opportunistically as live individuals sent to our lab for genetic analyses. Exact collection locations were recorded using a GPS (Garmin Legend, Garmin USA).

The approximate location of sampling sites in a survey of genotypic diversity of P. antipodarum in the western US. Areas in gray are HUCs (Hydrologic Unit Code of the USGS) with known P. antipodarum populations, according to http://www.esg.montana.edu/aim/mollusca/nzms/. Site labels are described in Table 1. Not shown are two sites in the Great Lakes (see Table 1)
Allozyme analysis
We obtained 562 samples from 18 sites in both the eastern and western US (Table 1). Samples were either snap frozen in liquid nitrogen, or kept alive until analysis. Samples were genotyped using six allozyme loci (PEP-D, IDH-1, PGM, 6PGD, MPI, AAT). We used cellulose acetate gel electrophoresis and well-established protocols; triploid genotypes were scored by asymmetric staining for loci and genotypes where alleles are sufficiently distinct (Dybdahl and Lively 1995, 1996, 1998; Fox et al. 1996). The identity of alleles and their relative migration rates on the gels are given in Appendix Table 3. Individuals sharing identical genotypes at all loci by definition had the same allozyme multilocus genotype (MLG), and were considered members of the same clonal lineage.
DNA extraction
For DNA analysis, samples were preserved in 95% EtOH and returned to the lab where they were transferred to 1 × TE buffered (10 mM Tris, 1 mM EDTA) 70% EtOH. Total genomic DNA was extracted using the chelex bead method (Walsh et al. 1991) by dissection and removal of a small piece of tissue from the foot. Individuals were given unique identification serial numbers and vouchers were preserved in buffered 70% EtOH. DNA extractions were used for either mitochondrial DNA sequencing, microsatellite DNA genotyping, or both. All of sampled individuals that were assayed for mtDNA haplotype were also genotyped using microsatellite markers.
Mitochondrial DNA sequencing
Sequences for the cytochrome b (cyt b) gene were obtained from up to 8 individuals from 22 sites for a total of 60 samples (Table 1; Fig. 1). (All of these individuals also were genotyped using microsatellite markers.) We used published primers and protocols from Neiman and Lively (2004) to amplify 431 bp of the cyt b gene. PCR fragments were cleaned using Shrimp Alkaline Phosotase (5 units) and Exonuclease I (0.5 units) by incubation at 30 min at 37°C, followed by 15 min at 80°C. Direct sequencing of cleaned PCR fragments was done using Big Dye 3.1 (ABI) chemistry on an ABI 3730 capillary sequencer. Forward and reverse sequences were proofread and aligned with Sequencher 4.2.2 (Genecodes). Sequence alignment was checked manually. COLLAPSE was used to identify unique haplotypes.
Microsatellite DNA amplification and analysis
Microsatellite amplification was attempted on at least 10 individuals from each sampled location, for a total of 322 samples from 20 sites (see Table 1). We used five of the published loci from Weetman et al. (2001): Pa254, Pa56, Pa112, Pa143, and Pa121. Each locus was amplified using ten uL PCR reactions with the following specific reagent concentrations: 10 mM Tris–HCl (pH 8.3), 50 mM KCl, 200 μM of each dNTP, 2 mM MgCl2, 0.5 μM of each primer, and 0.05 U of AmpliTaq (ABI). Our thermocycling parameters (modified from Weetman et al. 2001) were as follows: initial denaturation at 94°C for 60 s, followed by 30 cycles of 94°C for 30 s, 61°C for 30 s, and 72°C for 60 s, with a final extension at 72°C for 30 min. For each primer pair, the forward primer was labeled with fluorescent dye to facilitate automated fragment size analysis. Following amplification, PCR products were combined and diluted with size standard (Genescan 500 LIZ from ABI) and fragment size analysis was performed on an ABI 3730 capillary sequencer. We described allele size scoring bins in STRand (Hughes 1998) according to the microsatellite repeat motifs previously identified (Weetman et al. 2001). These predefined bins allowed STRand to match the size of a particular allele with an allele generated by the locus repeat motif. We focused our analysis of allele sizes on those within the size ranges previously identified by Weetman et al. (2001, 2002). Stutter peaks, caused by DNA strand slippage during PCR, were ignored. Peaks far outside the previously described allele size range were ignored because they could represent nonspecific amplification products. Our PCR protocols included a long extension reducing the presence of “plus A” peaks. Our microsatellite methods differ from those used in a recent survey of the Snake River in Idaho (Hershler et al. 2010). Their scoring methods are less detailed, but their results show that they considered much wider allele size ranges (over 50 repeats) than expected from the known allelic variation in microsatellites in P. antipodarum (Weetman et al. 2001, 2002).
Clonal P. antipodarum are triploid (Wallace 1992; Dybdahl and Lively 1995) which adds difficulty when determining heterozygous genotypes. It is straightforward to determine the genotype of individuals with one or three different allele sizes, but scoring individuals with only two different alleles is more difficult. Heterozygotes were determined by analyzing differences in peak height. An allele represented twice in the genotype (e.g., A in AAB) was expected to have a larger peak magnitude. However, complicating this process further, we expected shorter alleles to be amplified more compared to larger alleles, sometimes causing large allele drop-out (Selkoe and Toonen 2006). This sometimes results in short alleles having taller peak heights than longer alleles. If two clearly defined alleles with equal peak height were found in a sample, then we scored the longer peak (larger number of repeat units) as being represented twice in the genotype. For example, if peak A is 10 repeats long and B is 20 repeats and of equal height, then the genotype would be scored as ABB. Allele sizes were scored twice for each sample and any ambiguous samples were re-amplified and rescored. We compiled the five-locus MLG of the sampled individuals. Individuals that share alleles at all loci have the same MLG, and are considered to be members of a clonal lineage.
Combined multilocus genotypes
To measure genotypic diversity, we combined genotypic data from allozymes, mtDNA, and msDNA into a combined MLG. Conclusions about the association among markers within individuals in these combined MLG were confirmed in several ways. First, all of the individual samples taken from the field that were typed for mtDNA haplotype were also assayed for their multilocus microsatellite genotype. Second, although allozyme markers were assayed on a different set of individuals in most cases, populations were genotypically monomorphic for all three markers at all sites except S 20 and S 22 (Table 1). For the monomorphic sites, we concluded that all individuals with the single multilocus genotype at the DNA markers (either the mtDNA haplotype, the multilocus microsatellite genotype, or both) share the multilocus allozyme genotype found in sampled individuals from the same site. Third, for the genotypically diverse Bliss site (S 22), we genotyped the same individuals for all three markers (Table 1). Finally, for the genotypically diverse Malad River site (S 20), all individuals assayed for mtDNA were also assayed for their microsatellite genotype, confirming the association between these two markers. In addition, we ran an additional set of samples and we assayed these samples for all three markers.
Results
Allozyme MLG diversity
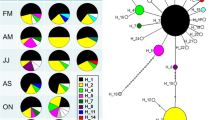
The samples of P. antipodarum in the eastern and western US recovered a total of 3 allozyme MLGs (Table 2). All populations sampled in the western US, with one exception, were comprised of individuals with the same MLG (referred to as US 1 in Table 2). The two sampled populations from the eastern US (Lakes Ontario and Superior) had a second MLG (see US 2 in Table 2). A single site in the middle Snake River near Bliss, ID (site S 22), was comprised of a mix of two different allozyme MLGs (see US 1 and US 3 in Table 2).
Mitochondrial DNA sequence diversity
The samples across the US range returned three cytochrome b haplotypes (Table 2). All except one western US population were comprised of individuals with a single haplotype (referred to as H06). This haplotype is identical to haplotype 37 (genbank AY570216) from Neiman and Lively (2004). The Bliss population (site S 22) also contained a second divergent haplotype H19 (1.9% uncorrected sequence divergence from the H06 haplotype; genbank HG680431). The third haplotype, referred to as H05 and similar to haplotype H19 (less than 0.2% uncorrected sequence divergence), was found exclusively in samples from the Great Lakes (sites E1 and E2). This haplotype is identical to haplotype 22 (genbank AY570201) from Neiman and Lively (2004).
Microsatellite DNA MLG diversity
Analysis of the 372 individuals across the 20 sites returned four different msDNA MLGs (Table 2). All western US populations except two were comprised of a single msDNA MLG (see US 1 in Table 2). A second msDNA MLG was present in one western US location (S 20: Malad River, ID) at a moderate frequency (15%) (see US 1a in Table 2). A third msDNA MLG was found at the Bliss, ID, site (S 22) (see US 3 in Table 2). Finally, a fourth msDNA MLG was found in Great Lakes sites (see US 2 in Table 2).
Total genotypic diversity
The combined genotypic data from allozymes, mtDNA, and msDNA revealed a total of four different genotypes, one of which was widespread in the western US (Table 2). The combined MLG are numbered in Table 2. The three different allozyme genotypes were each associated with a distinct mtDNA haplotype and msDNA MLG, and these clones were designated as US 1, US 2, and US 3. Individuals designated as US 1a have a distinct combined msDNA and allozyme MLG, but share their mtDNA haplotype (H06) with US 1 (Table 2).
Discussion
We surveyed three types of genetic markers in US populations of P. antipodarum to determine the genotypic diversity of a highly invasive clonal species. The combined genetic marker data showed that a single MLG (US 1) occupies the vast majority of the range in the western US. In addition, a single distinct MLG (US 2) was identified in the Great Lakes (Ontario and Superior). These results indicate that two different broadly distributed clonal populations in the western US and the Great Lakes were each comprised of individuals that are derived from a single maternal lineage (US 1 and US 2). This survey also detected two clones (US 1a and US 3) that are distributed narrowly in the middle Snake River of Idaho. Hence, genetic markers, including highly variable microsatellite DNA, show that expansion across a broad geographic range by P. antipodarum has occurred despite the near absence of genotypic variation within and among populations.
Propagule pressure has the potential to result in greater levels of genetic diversity than found in the native range (Lockwood et al. 2005; Kolbe et al. 2004; Lavergne and Molofsky 2007; Facon et al. 2008). In contrast, the importance of propagule pressure in building genetic diversity in US invasive populations of P. antipodarum appears to be minimal. Our results suggest that a total of four clonal lineages (US 1, US 1a, US 2, US 3) occur in the US, three of which occur only in the western US (US 1, US 1a, US 3). The US 2 clone from the Great Lakes samples is distinct from western US clones at all three genetic markers. The mtDNA haplotype of US 2 is identical to that of the EU A clone that is widespread across continental Europe (Stadler et al. 2005). Two of the three clones in the western US (US 1 and US 3) appear to represent introductions from distinct clonal lineages because they differ from each other at all three genetic markers. According to their mtDNA haplotype, US 1 and US 3 also come from highly divergent clades (Neiman and Lively 2004). The third clone in the Snake River area (US 1a) has a novel msDNA genotype that might represent either introduced variation, or a newly arisen lineage from US 1 that differs only by recent mutation. US 1 and US 1a share alleles for four of the five loci that were scored (Pa 56, 112, 121, 143) (Table 2). Mutational variants at three of these loci represent losses/gains of at least 2 repeats. This level of mutational change is consistent with previous studies of this species in Great Britain (Weetman et al. 2002). However, variation between US 1 and US 1a at Pa 121 represents the gain/loss of at least 15 repeats, and this exceeds the level of mutational gain/loss in Great Britain (at most 4 repeats as reported in Weetman et al. 2002). Therefore, it seems likely that the US 1a genotype represents a third introduced genotype in the western US rather than newly arisen mutation variation.
Studies of microsatellite mutational processes and allelic variation on introduction populations of P. antipodarum in Great Britain uncovered a large number of microsatellite MLGs. A survey of eight clonal populations across Great Britain using four of the same microsatellite markers and the same allele size range that we used revealed a total of 34 MLGs, of which four were common and one was widespread (Weetman et al. 2002). Weetman et al. concluded that the microsatellite variation resulted from mutational input within Great Britain populations which were established no later than the 1880′s. Consistent with the mutational input explanation, the most widespread genotype in Great Britain was likely the ancestor of 16 MLGs that differ by only a few mutational steps (Weetman et al. 2002). Given the shorter time in the western US populations since introduction for mutational input, it is not surprising that our survey revealed different microsatellite MLGs only in individuals that also differed in mtDNA or allozyme genotype. In contrast, a microsatellites survey of Snake River, ID, populations, Hershler et al. (2010) scored more microsatellite alleles and MLGs. However, unlike our study, they used a range of allele sizes much larger than would be consistent with the current understanding of microsatellite variation in P. antipodarum (Weetman et al. 2001, 2002). Their study also did not consider other genetic and phenotypic markers of clone identity to support their conclusions.
Of the three clonal lineages in the western US (US 1, US 1a, US 3), only the US 1 clone was found in all populations. Similarly, continental European populations of P. andipodarum are comprised of a single MLG (Ponder 1988, Hauser et al. 1992). In the western US, US 1a and US 3 were restricted to two of our collection sites in the middle Snake River, ID. These sites are near the oldest known records of P. antipodarum in the US and the presumed original introduction (Proctor et al. 2007). It is not clear why the US 1a and US 3 genotypes have a restricted range. One possibility is that they might have been introduced together with US 1, but have not expanded. If so, then perhaps US 1a and US 3 differ ecologically from US 1 in a way that limits their capacity to occupy new environments. Alternatively, they might have been introduced more recently and have not yet had sufficient time to disperse. If so, and if these genotypes differ ecologically from US 1 in a way that enhances their capacity to occupy new environments, then they might represent the potential for expansion in habitats not currently occupied by US 1. Such cryptic secondary invasions of specific genotypes can sometimes be more ecologically harmful than established genotypes (Saltonstall 2002).
Either evolutionary adaptation or generalism and plasticity are required when invasive species expand across a range of novel environments. A growing number of studies suggest that evolutionary adaptation facilitates range in invasive species (Maron et al. 2004; Huey et al. 2000; Lee et al. 2003, 2007; Muller-Scharer et al. 2004; Bossdorf et al. 2005; Lavergne and Molofsky 2007). Expansion by adaptation is much faster if there is sufficient genetic variation within the invading population (Garcia-Ramos and Rodriguez 2002). If evolved adaptation is important for invasion, then there is a paradox: why are bottlenecked invasive species successful even though bottlenecked native species often are susceptible to extinction (Frankham 2005; Roman and Darling 2007)? Furthermore, if evolvability is important for invasions, then why are self-fertilization and clonal reproduction over-represented among invasive species (Barrett et al. 2008; Roman and Darling 2007)?
In clonal populations of P. antipodarum in the native range, abundant genotypic variation and restricted ecological distribution of individual genotypes suggest that niche evolution occurs through clonal selection (Fox et al. 1996; Jokela et al. 1999; Jokela et al. 2003). Individual clones are strongly segregated among lakes (Dybdahl and Lively 1995) and among different lake-bottom habitats in two New Zealand lakes. Hence, it is surprising that the western US and European populations of P. antipodarum have expanded across a wide array of niches without the degree of clonal diversity found in the native range. In the western US, the range includes habitats as diverse as coastal estuaries and high elevation streams (Dybdahl and Kane 2005). Thus, like other species with uniparental reproduction that undergo bottlenecks and lose variation during invasion (Roman and Darling 2007), it remains an open question how populations that lack genetic diversity can be successful across a wide environmental gradient (see also Mergeay et al. 2006; Tepolt et al. 2009).
In P. antipodarum, evolved adaptation to local conditions in the western US might be possible if genetic variation has accumulated from mutational input within an individual clonal lineage such as US 1. Even though our survey did not detect genotypic variation at neutral genetic marker loci, populations of P. antipodarum from the Columbia and Snake Rivers have low but detectable levels of quantitative genetic variation for growth rate and reproduction (Dybdahl and Kane 2005). Nevertheless, evolved responses might be limited by low levels of quantitative genetic variation, in addition to the absence of genotypic diversity that could fuel clonal selection in invasive P. antipodarum populations.
Rather than adaptive contemporary evolution, P. antipodarum might be able to occupy a range of niches because they maintain high fitness over a broad range of environments via phenotypic plasticity (Richards et al. 2006). Such general-purpose genotypes have long been assumed to be associated with invasiveness (Baker 1965). There is little indication that general-purpose genotypes exist in populations of P. antipodarum in the native range (Dybdahl and Lively 1995; Jokela et al. 2003), although it is possible that plasticity and generalism have evolved during colonization or expansion (see Drown et al. 2010). Understanding the form of plasticity that is likely to have evolved during the process of invasion may help to explain the paradox of how an invasive species can become broadly successful despite a lack of genetic diversity.
References
Amsellam L, Noyer JL, Le Bourgeois T, Hossaert-McKey M (2000) Comparison of genetic diversity in the invasive weed Rubus alceifolius Poir. (Rosaceae) in its native range and in areas of introduction, using amplified fragment length polymorphism (AFLP) markers. Mol Ecol 9:443–455
Baker HG (1965) Characteristics and modes of origin of weeds. In: Baker HG, Stebbins GL (eds) The genetics of colonizing species. Academic Press, New York, pp 147–168
Barrett SCH, Colautti RJ, Eckert CC (2008) Plant reproductive systems and evolution during biological invasion. Mol Ecol 17:373–383
Bossdorf O, Auge H, Lafuma L, Rogers WE, Siemann E, Prati D (2005) Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia 144:1–11
DeWalt SJ, Hamrick JL (2004) Genetic variation of introduced Hawaiian and native Costa Rican populations of an invasive tropical shrub, Clidemia hirta (Melastomataceae). Am J Bot 91:1155–1162
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Drown DM, Levri EP, Dybdahl MF (2010) Invasive genotypes are opportunistic specialists not general purpose genotypes. Evol Appl (in press)
Dybdahl MF, Kane SL (2005) Adaptation versus environmental tolerance in the success of a clonal invader. Ecology 86:1592–1601
Dybdahl MF, Lively CM (1995) Diverse, endemic, and polyphyletic clones in mixed populations of a freshwater snail Potamopyrgus antipodarum. J Evol Biol 8:385–398
Dybdahl MF, Lively CM (1996) The geography of coevolution: comparative population structures for a snail and its trematode parasite. Evolution 50:2264–2275
Dybdahl MF, Lively CM (1998) Host-parasite coevolution: evidence for rare advantage and time-lagged selection in a natural population. Evolution 52:1057–1066
Facon B, Pointier J, Jarne P, Sarda V, David P (2008) High genetic variance in life-history strategies within invasive populations by way of multiple introductions. Curr Biol 18:363–367
Fox JA, Dybdahl MF, Jokela J, Lively CM (1996) Genetic structure of coexisting sexual and clonal subpopulations in a freshwater snail (Potamopyrgus antipodarum). Evolution 50:1541–1548
Frankham R (2005) Resolving the genetic paradox of invasive species. Heredity 94:385
Garcia-Ramos G, Rodriguez D (2002) Evolutionary speed of species invasion. Evolution 56:661–668
Grapputo A, Boman S, Lindstrom L, Lyytinen A, Mappes J (2005) The voyage of an invasive species across continents: genetic diversity of North American and European Colorado potato beetle populations. Mol Ecol 14:4207–4219
Hall RO, Tank J, Dybdahl MF (2003) Exotic snails dominate nitrogen and carbon cycling in a highly productive stream. Front Ecol Environ 1:407–411
Hall RO, Dybdahl MF, Vanderloop M (2006) Invasive species and energy flow: extremely high secondary production of introduced snails in rivers. Ecol Appl 16:1121–1131
Hauser L, Carvalho GR, Hughes RN, Carter RE (1992) Clonal structure of the introduced freshwater snail Potamopyrgus antipodarum (Prosobranchia: Hydrobiidae), as revealed by DNA fingerprinting. Proc R Soc Lond 249:19–25
Hershler R, Liu HP, Clark WH (2010) Microsatellite evidence of invasion and rapid spread of divergent New Zealand mudsnail (Potamopyrgus antipodarum) clones in the Snake River basin, Idaho, USA. Biol Invasions 12:1521–1532
Huey RB, Gilchrist GW, Carlson ML, Berrigan D, Serra L (2000) Rapid evolution of a geographic cline in size in an introduced fly. Science 287:308–309
Hughes S (1998) STRand nucleic acid analysis software. Regents of the University of California, Davis (http://www.vgl.uc-davis.edu/STRand)
Jokela J, Dybdahl MF, Lively CM (1999) Habitat-specific variation in life-history traits, clonal population structure, and parasitism in a freshwater snail (Potamopyrgus antipodarum). J Evol Biol 12:350–360
Jokela J, Lively CM, Dybdahl MF, Fox. JA (2003) Genetic variation in sexual and clonal lineages of a freshwater snail. Biol J Linnean Soc 79:165–181
Kerans BL, Dybdahl MF, Gangloff MM, Jannot JE (2005) Macroinvertebrate assemblages and the New Zealand mud snail, a recent invader to streams of the Greater Yellowstone Ecosystem. J North Am Benthol Soc 24:123–138
Kolbe JJ et al (2004) Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181
Lavergne S, Molofsky J (2007) Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc Natl Acad Sci 104:3883–3888
Lee CE (2002) Evolutionary genetics of invasive species. Trends Ecol Evol 17(8):386–391
Lee CE, Remfert JL, Gelembiuk GW (2003) Evolution of physiological tolerance and performance during freshwater invasions. Integr Comp Biol 43:439–449
Lee CE, Remfert JL, Chang Y-M (2007) Response to selection and evolvability of invasive populations. Genetica 129:179–192
Lee CE, Gelembiuk GW (2008) Evolutionary origins of invasive populations. Evol Appl 1:427–448
Lockwood JL, Casey P, Blackburn T (2005) The role of propagule pressure in explaining species invasions. Trends Ecol Evol 20:223–228
Maron JL, Vila` M, Bommarco R, Elmendorf S, Beardsley P (2004) Rapid evolution of an invasive plant. Ecol Monogr 74: 261–280
Mergeay J, Verschuren D, De Meester L (2006) Invasion of an asexual water flea clone throughout Africa and rapid displacement of a native sibling species. Proc R Soc B 273:2839–2844
Muller-Scharer H, Schafner U, Steinger T (2004) Evolution in invasive plants: implications for biological control. Trends Ecol Evol 19:417–422
Neiman M, Lively CM (2004) Pleistocene glaciation is implicated in the phylogeographical structure of Potamopyrgus antipodarum, a New Zealand snail. Mol Ecol 13:3085–3098
Ponder WF (1988) Potamopyrgus antipodarum, a molluscan colonizer of Europe and Australia. J Mulluscan Stud 54:271–286
Proctor T, Kerans B, Clancey P, Ryce E, Dybdahl M, Gustafson D, Hall R, Pickett F, Richards D, Waldeck RD, Chapman J, Wiltshire RH, Becker D, Anderson M, Pitman B, Lassuy D, Heimowitz P, Dwyer P, Levri EP (2007) National management and control plan for the New Zealand Mudsnail (Potamopyrgus antipodarum). US Fish and Wildlife Service Report. www.anstaskforce.gov/Documents/NZMS_MgmtControl_Final.pdf Accessed 27 July 2010
Richards CL, Bossdorf O, Muth NZ, Gurevitch J, Pigliucci M (2006) Jack of all trades, master of some? On the role of phenotypic plasticity in plant invasions. Ecol Lett 9:981–993
Roman J, Darling JA (2007) Paradox lost: genetic diversity and the success of aquatic invasions. Trends Ecol Evol 22:454–464
Saltonstall K (2002) Cryptic invasion by a non-native genotype of the common reed, Phragmites australis, into North America. Proc Natl Acad Sci 99:2445–2449
Selkoe KA, Toonen RJ (2006) Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9(5):615–629
Stadler T, Frye M, Neiman M, Lively CM (2005) Mitochondrial haplotypes and the New Zealand origin of clonal European Potamopyrgus, an invasive aquatic snail. Mol Ecol 14(8):2465–2473
Tepolt CK, Darling JA, Bagley MJ, Geller JB, Blum MJ, Grosholz ED (2009) European green crabs (Carcinus maenas) in the northeast Pacific: genetic evidence for high population connectivity and current-mediated expansion from a single introduced source population. Divers Distrib 15:997–1005
Wallace C (1992) Parthenogenesis, sex and chromosomes in Potamopyrgus. J Molluscan Stud 58:93–107
Walsh PS, Metzger DA, Higuchi R (1991) Chelex-100 as a medium for simple extraction of DNA for PCR based typing from forensic material. Biotechniques 10:506–513
Weetman D, Hauser L, Carvalho GR (2001) Isolation and characterization of di- and trinucleotide microsatellites in the freshwater snail Potamopyrgus antipodarum. Mol Ecol Notes 1:185–187
Weetman D, Hauser L, Carvalho GR (2002) Reconstruction of microsatellite mutation history reveals a strong and consistent deletion bias in invasive clonal snails, Potamopyrgus antipodarum. Genetics 162:813–822
Zaranko DT, Farara DG, Thompson FG (1997) Another exotic Mollusk in the Laurentian Great Lakes: the New Zealand native Potamopyrgus antipodarum (Gray 1843) (Gastropoda, Hydrobiidae). Can J Fish Aquat Sci 54:809–814
Acknowledgments
We would like to thank Angie Kotchetova, Kendra Lyons, Jennifer Thorson, and Jonathan Finger for help with molecular genotyping, and people too numerous to mention for collecting and sending specimens. Funding was provided by NSF DEB 0296049 and the US Fish and Wildlife Service.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dybdahl, M.F., Drown, D.M. The absence of genotypic diversity in a successful parthenogenetic invader. Biol Invasions 13, 1663–1672 (2011). https://doi.org/10.1007/s10530-010-9923-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-010-9923-4