
Abstract
Mytilopsis sallei is one of a small number of tropical estuarine organisms known to have successfully invaded habitats outside their native range in the Caribbean. This bivalve now occurs in several major ports in East Asia, which suggests the transport of larvae and/or adults by vessels. However, little work has been done to determine transfer pathways because direct evidence is difficult to obtain. Here we test whether there is sufficient genetic variability in a mitochondrial marker of established Asian populations of M. sallei to allow for future reconstruction of invasion history. We sampled a 376-base-pair fragment of the mitochondrial cytochrome oxidase I (COI) of M. sallei for 254 individuals representing 11 populations from Singapore, India, Hong Kong and Taiwan. We found high variability with 24 positions distinguishing 15 haplotypes. Haplotype diversity ranged between 0.6 and 0.8 in eight Singapore populations, and an analysis of molecular variance showed that there was no significant genetic segregation in these populations. Observed haplotype diversity was also high in a population from Visakhapatnam, India, but was slightly lower in samples from Taiwan and Hong Kong. Preliminary data also indicate that Singapore, India, Hong Kong, and Taiwan populations may have different dominant haplotypes. These results suggest that there is sufficient genetic variability to use mitochondrial markers for reconstructing the invasion history of Mytilopsis sallei, when larger sample sizes become available.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There are only a small number of documented cases of marine invasions by invertebrates in tropical coastal areas (Crisci et al. 2003; Hutchings et al. 2002; Lewis et al. 2006; but see Carlton and Eldredge 2009). Arguably the best known is the establishment of the estuarine Caribbean dreissenid bivalve Mytilopsis sallei (Récluz) in and near major international ports and marinas in Australasia (e.g., Furuse and Hasegawa 1984; Karande and Menon 1975; Morton 1981, 1989; Nuttall 1990a, b; Stepien et al. 2001; Tan and Morton 2006; Wangkulangkul and Lheknim 2008, as M. adamsi; Willan et al. 2000) and the Mediterranean (Galil and Bogi 2009). Mytilopsis sallei is a highly adaptable New World relative of the freshwater Asian zebra mussel Dreissena polymorpha (Pallas), the latter being on the top 100 list of the world’s worst invasive alien species as classified by the Global Invasive Species Database (see http://www.issg.org/). Given their ecological resemblance, M. sallei is likely to become similarly notorious because it is capable of surviving in a wide range of salinities (Ramachandra Raju et al. 1975) and temperatures (Tan, unpubl. obs.). The dispersal of this versatile and fecund species from the Caribbean to Southeast Asia has generally been attributed to shipping, either by way of the Atlantic to West Africa (Oliver et al. 1998) or via the Pacific through the Panama Canal (Morton 1981). The most likely modes of transportation are in cargo vessel ballast water (Chu et al. 1997; Williams et al. 1988) and/or through hull fouling (Willan et al. 2000). Unfortunately, a direct identification of the pathways and frequencies of invasion would require detailed and frequent examination of ballast water and ship hulls that are difficult to sample adequately, even in the best of circumstances. A potential alternative technique is to develop genetic markers that can identify invasive populations from different locations. This approach can potentially also determine the origin, direction and intensity of gene flow (Avise 2000; Chandler et al. 2008; Wang et al. 2009, 1999; Zardus and Hadfield 2005). Using these techniques for M. sallei is currently not feasible because too few sequences are available and it is thus unknown whether the species harbours sufficient genetic variability for reconstructing invasion pathways. Here we describe the genetic variability of the mitochondrial cytochrome c oxidase subunit I (COI) of M. sallei from established populations in Singapore, India, Hong Kong, and Taiwan.
Materials and methods
Sampling
To assess the extent of genetic variation of Mytilopsis across different spatial scales, eight populations in Singapore (Kranji, Sembawang, Punggol, Serangoon, Potong Pasir, Geylang, Kim Seng, Pandan) and three populations elsewhere in South and East Asia (Visakhapatnam on the east coast of India, Tolo Harbour in Hong Kong, and Kaohsiung in Taiwan) were sampled (see Fig. 1). Unfortunately, several attempts to obtain specimens from its native Caribbean region were unsuccessful. Animals were collected by hand during low tide in the period between July 2004 and March 2005. Prior to tissue extraction, live individuals were preserved immediately in 100% ethanol for at least a few days with their valves held apart. Between 20 and 28 animals from each site were used for mtDNA extraction and subsequent analyses. As initial tests failed to detect sex-linked haplotype specificity (Geller 1994, 1996; Stepien et al. 1999), no attempt was made to distinguish sexes in samples.

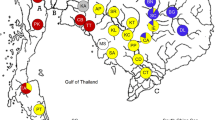
Sampling localities and composition of COI haplotypes among (A) East Asian (India, Hong Kong and Taiwan) and (B) Singapore populations of Mytilopsis sallei. Pie charts indicate haplotype proportions. In (B), pie charts are centred approximately over actual collecting locations, except Geylang
mtDNA extraction, purification and amplification
DNA was extracted from posterior mantle tissue using a commercial kit (DNEasy, Qiagen) according to manufacturer’s instructions. Extracted genomic DNA (1–2 μl) was initially subjected to the polymerase chain reaction (PCR) using COI (cytochrome c oxidase I) universal primers LCO1490 and HCO2198 (Folmer et al. 1994). Sequence quality was dissatisfactory and new primers internal to the universal primers were designed (forward: 5′-GGAGCTTAGTGCTCCTGGA-3′ and reverse: 5′AAGCATTGTCAGCCCACCA-3′). These primers amplified a ~492-base pair (bp) fragment of the COI gene. Each 25 μL reaction comprised 2.5 μL 10 × PCR buffer (Qiagen), 1.2 μL each of the oligonucleotide primers (10 μM), 1 μL dNTP’s (0.2 mM), 0.5 U Taq polymerase (Takara Ex Taq Hotstart version) and autoclaved distilled water to volume. The thermal profile used an initial denaturation cycle at 95°C for 3 min followed by 35 cycles of 94°C for 1 min, annealing at 57°C (30 s), and extension at 72°C (90 s). A 2 min extension at 72°C was added at the end of the cycle to increase copy number. After verification via gel electrophoresis, the PCR products were purified using QIAquick PCR Purification Kit (Qiagen) and cycle-sequenced twice in the reverse direction using the Big Dye Terminator version 3.1 cycle sequencing kit (Applied Biosystems). The cycle-sequenced product underwent a final purification step using CleanSEQ Dye Terminator Removal kit (Agencourt Bioscience Corporation). Sequencing with the new primers was carried out on an ABI Prism 3,100 Genetic Analyzer at the Department of Biological Sciences, National University of Singapore. Sequences representing the different haplotypes were deposited in GenBank (accession numbers DQ078480–078494).
Analysis
Sequences were contigued with SEQUENCHER 3.1 (Gene Codes) and manually edited before creating consensus sequences. The alignment of the consensus sequences was indel-free and translatable into amino acids. Haplotype diversity (h) was estimated according to Nei (1987) and analyses of molecular variance (AMOVA) were performed for the relatively well sampled Singaporean populations using ARLEQUIN 2.0 (Excoffier et al. 1992; Schneider et al. 2000) to determine if any genetic structure can be detected. Pairwise ϕSTs were employed to provide preliminary estimates of genetic distances between sampled populations.
Results
DNA from 254 individuals in Singapore, India, Hong Kong and Taiwan were extracted, amplified and sequenced. A total of 15 haplotypes with variation across 24 nucleotide positions of the 376-bp COI gene fragment were identified. Most base changes occurred in the third position and were silent. Changes in the first and second positions were rare but resulted in amino-acid changes. Fourteen haplotypes were observed in Singapore, six in India and Hong Kong, and four in Taiwan (Table 1). Between four and eight haplotypes were obtained from the 20–28 individuals sampled at each location (Table 1). Four haplotypes (the lowest number) were obtained from a population in Kaohsiung, Taiwan, whilst twice the number of haplotypes was observed in a population sampled from Kranji, Singapore (see also Fig. 1).
With one exception all common haplotypes (found in >5 specimens) occurred in at least two countries. At each site in Singapore, the number of haplotypes varied between five and eight. Haplotypes 1 and 11 were dominant in Singapore and Taiwan, but were also found in all other East Asian populations. Haplotypes 1 and 11 occurred in relative proportions of up to 70% (Table 1), and contributed between 48 and 86% at all localities. In the majority of cases, haplotype 1 was more common than haplotype 11, except for the samples from Tolo Harbour (Hong Kong), Visakhapatnam (India) and Geylang (Singapore) where the reverse proportion was observed. Haplotype 6 was also present in nearly all localities except for two Singapore sites. About half the haplotypes sequenced are relatively rare (10% or less). We also found four unique haplotypes (nos 3, 7, 9 and 12), i.e., haplotypes represented by a single individual, in populations from Singapore and India (Table 1). Haplotype diversity (h) ranged between 0.518 and 0.790, while nucleotide diversity (π) was between 0.00522 and 0.00988 (Table 2). In general, populations from Singapore and India had higher genetic diversity than those from Hong Kong and Taiwan.
The 15 haplotypes were relatively diverse, with uncorrected genetic distances ranging between 0.27 and 3.19%. The maximum genetic distance recorded was between haplotypes 3 and 10 (Table 3). An AMOVA showed that there was little or no genetic structure among the eight Singapore populations, i.e., 99% of the variation was accounted for within each population (P = 0.196, df = 184). However, pairwise analyses of pooled Singapore samples with populations from Taiwan, Hong Kong and India revealed significant differences (P < 0.05) between those of Singapore and Hong Kong, and those of Singapore and India (Table 4). There was no significant difference between pooled Singapore and Taiwan populations (Table 4).
Discussion
In this study we identified a surprisingly large number of different haplotypes (15) that are distinguished by 24 variable sites within a relatively short 376 bp segment of COI gene. This variability was found in 254 individuals sampled from Singapore, India, Hong Kong, and Taiwan. This suggests that the gene has sufficient genetic variability for studying invasion history and gene flow in Mytilopsis sallei. At least four haplotypes were identified from each of the 11 populations sampled. Within Singapore, Mytilopsis populations had a total of 14 different haplotypes in eight populations located not more than 15 km apart, i.e., gene flow between the populations appear unstructured, which may indicate that gene exchange occurs among the populations either naturally or assisted by anthropogenic shipping activities. Interpreting the comparative data for Singapore, India, Hong Kong, and Taiwan is more difficult. Despite several attempts, we were not able to obtain samples from the native range, and the relatively small number of individuals available for the populations from India, Hong Kong, and Taiwan precluded a more robust analysis. Nevertheless, our preliminary data suggest that Mytilopsis populations in India and Hong Kong have different dominant haplotypes from those in Singapore and Taiwan. Whilst high genetic diversity is not unexpected for invasive species (Stepien et al. 2005) and marine organisms (Dupont et al. 2003; Crandall et al. 2008), the very high genetic variability here observed for invasive Mytilopsis populations is surprising. The data can either be explained by a small number of invasions through a genetically unusually variable population or the high genetic diversity is due to multiple invasions. We favour the latter explanation given that high diversity is found in four countries and it appears unlikely that single invasions would consistently produce such a pattern. Multiple founders and/or multiple founding sources have also been observed for the Chinese mitten crab Eriocheir sinensis that is now established in Europe (Wang et al. 2009), and the European green crab Carcinus maenas in Canada (Roman 2006). This is in contrast to the marine gastropod Rapana venosa, which lacks genetic diversity in introduced populations compared to those in the native range (Chandler et al. 2008).
The maximum haplotype diversity for the COI gene, h = 0.79, was recorded for the Kranji-Singapore population. This haplotypic diversity is significantly higher than reported for populations of other surface-dwelling dreissenoid relatives such as Congeria kusceri (h = 0.66; Stepien et al. 2001). The sequence divergence of COI within Mytilopsis sallei populations yielded a range of genetic distance from 0.276 to 3.19%. Intraspecific COI variability in M. sallei was higher than in other members of the Dreissenidae (up to 1.1%; Quaglia et al. 2008; Therriault et al. 2004). Given their high genetic variability, there may be inherent difficulties in controlling invasive populations of these bivalves. However, since this study was carried out, a barrage built across Marina estuary in 2009 has effectively isolated what was perhaps the largest Mytilopsis community (including Kim Seng, Potong Pasir and Geylang populations sampled in this study) in Singapore from the sea. It remains to be seen if these genetically variable populations can survive in what will eventually be a freshwater reservoir fed by rain and urban runoff.
Our study unfortunately also demonstrates why the use of gene sequences for reconstructing the invasion history of species faces serious sampling issues when source and invasive populations are found in disparate geographic areas. Obtaining samples from all relevant areas is very expensive and time-consuming and the number of samples that needs to be analysed remains unknown until a preliminary assessment of the genetic variability within the populations is completed. In the case of M. sallei, our preliminary assessment revealed that the COI gene has sufficient variability for reconstructing the invasion history of this species. In addition, we have also shown that a large number of individuals and locations need to be sampled in order to obtain a good coverage of haplotype diversity.
References
Avise JC (2000) Phylogeography. The history and formation of species. Harvard University Press, Cambridge, p 447
Carlton JT, Eldredge LG (2009) Marine bioinvasions of hawai’i. Bish Mus Bull Cult Environ Stud 4:1–202
Chandler EA, McDowell JR, Graves JE (2008) Genetically monomorphic invasive populations of the rapa whelk, Rapana venosa. Mol Ecol 17:4079–4091
Chu KH, Tam PF, Fung CH, Chen QC (1997) A biological survey of ballast water in container ships entering Hong Kong. Hydrobiologia 352:201–206
Crandall ED, Jones ME, Muñoz MM, Akinronbi B, Erdmann MV, Barber PH (2008) Comparative phylogeography of two seastars and their ectosymbionts within the Coral Triangle. Mol Ecol 17:5276–5290
Crisci JV, Katinas L, Posadas P (2003) Historical biogeography. An introduction. Harvard University Press, Cambridge, p 250
Dupont L, Jollivet D, Viard F (2003) High genetic diversity and ephemeral drift effects in a successful introduced mollusc (Crepidula fornicata: Gastropoda). Mar Ecol Prog Ser 253:183–195
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:45479–45491
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Furuse K, Hasegawa K (1984) Mytilopsis sallei found in Tokyo Bay. Chiribotan 15:18
Galil BS, Bogi C (2009) Mytilopsis sallei (Mollusca: Bivalvia: Dreissenidae) established on the Mediterranean coast of Israel. Mar Biodiv Rec 2:1–4
Geller JB (1994) Sex-specific mitochondrial DNA haplotypes and heteroplasmy in Mytilus trossulus and Mytilus galloprovincialis populations. Mol Mar Biol Biotech 3:334–337
Geller J (1996) Molecular approaches to the study of marine biological invasions. In: Ferraris JD, Palumbi SR (eds) Molecular zoology: advances, strategies, and protocols. Wiley, New York, pp 119–132
Hutchings PA, Hilliard RW, Coles SL (2002) Species introductions and potential for marine pest invasions into tropical marine communities, with special reference to the Indo-Pacific. Pac Sci 56:223–233
Karande AA, Menon KB (1975) Mytilopsis sallei, a fresh immigrant in Indian harbours. Bull Dept Mar Sci Univ Cochin 7:455–466
Lewis JA, Watson C, Ten Hove HA (2006) Establishment of the Caribbean serpulid tubeworm Hydroides sanctaecrucis Krøyer [in] Mörch, 1863, in northern Australia. Biol Inv 8:665–671
Morton B (1981) The biology and functional morphology of Mytilopsis sallei (Récluz) (Bivalvia: Dreissenacea) fouling Visakhapatnam harbour, Andhra Pradesh, India. J Moll Stud 47:25–42
Morton B (1989) Life-history characteristics and sexual strategy of Mytilopsis sallei (Bivalvia: Dreissenacea), introduced into Hong Kong. J Zool Lond 219:469–485
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York, p 512
Nuttall CP (1990a) A review of the Tertiary non-marine molluscan faunas of the Pebasian and other inland basins of north-western South America. Bull Brit Mus (Nat Hist) (Geol) 45:165–371
Nuttall CP (1990b) Review of the Caenozoic heterodont bivalve superfamily Dreissenacea. Palaeontology 33:707–737
Oliver PG, Holmes AM, Mettam C (1998) Mytilopsis leucophaeta (Conrad, 1831) (Bivalvia: Dreissenoidea), a species new to the British fauna. J Conch 36:13–18
Quaglia F, Lattuada L, Mantecca P, Bacchetta R (2008) Zebra mussels in Italy: where do they come from? Biol Inv 10:555–560
Ramachandra Raju P, Mangapathi Rao K, Ganti SS, Kalyanasundaram N (1975) Effect of extreme salinity conditions on the survival of Mytilopsis sallei (Pelecypoda). Hydrobiologia 46:199–206
Roman J (2006) Diluting the founder effect: cryptic invasions expand a marine invader’s range. Proc R Soc B 273:2453–2459
Schneider S, Roessli D, Excoffier L (2000) ARLEQUIN (version 2.0): a software for population genetics data analysis. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva, Geneva
Stepien CA, Hubers AN, Skidmore JL (1999) Diagnostic genetic markers and evolutionary relationships among invasive dreissenoid and corbiculoid bivalves in North America: phylogenetic signal from mitochondrial 16S rDNA. Mol Phylogenet Evol 13:31–49
Stepien CA, Morton B, Dabrowska KA, Guarnera RA, Radja T, Radja B (2001) Genetic diversity and evolutionary relationships of the troglodytic ‘living fossil’ Congeria kusceri (Bivalvia: Dreissenidae). Mol Ecol 10:1873–1879
Stepien CA, Brown JE, Neilson ME, Tumeo MA (2005) Genetic diversity of invasive species in the Great Lakes versus their Eurasian source populations: insights for risk analysis. Risk Anal 25:1043–1060
Tan KS, Morton B (2006) The invasive Caribbean bivalve Mytilopsis sallei (Dreissenidae) introduced to Singapore and Johor Bahru, Malaysia. Raffles Bull Zool 54:429–434
Therriault TW, Docker MF, Orlova MI, Heath DD, MacIsaac HJ (2004) Molecular resolution of the family Dreissenidae (Mollusca: Bivalvia) with emphasis on Ponto-Caspian species, including first report of Mytilopsis leucophaeta in the Black Sea basin. Mol Phylogenet Evol 30:479–489
Wang JJ, Huang ZG, Zheng CX, Lin N (1999) Population dynamics and structure of alien species Mytilopsis sallei in Fujian, China. J Oceanogr Taiwan Strait 18:372–377
Wang C, Li S, Fu C, Gong X, Huang L, Song X, Zhao Y (2009) Molecular genetic structure and evolution in native and colonized populations of the Chinese mitten crab, Eriocheir sinensis. Biol Inv 11:389–399
Wangkulangkul K, Lheknim V (2008) The occurrence of an invasive alien mussel Mytilopsis adamsi Morrison, 1946 (Bivalvia: Dreissenidae) in estuaries and lagoons of the lower south of the Gulf of Thailand with comments on their establishment. Aquat Invasions 3:325–330
Willan RC, Russell BC, Murfet NB, Moore KL, McEnnulty FR, Horner SK, Hewitt CL, Dally GM, Campbell ML, Bourke ST (2000) Outbreak of Mytilopsis sallei (Récluz, 1849) (Bivalvia: Dreissenidae) in Australia. Moll Res 20:25–30
Williams RJ, Griffiths FB, Van der Wal EJ, Kelly J (1988) Cargo vessel ballast water as a vector for the transport of non-indigenous marine species. Estuar Coast Shelf Sci 26:409–420
Zardus JD, Hadfield MG (2005) Multiple origins and incursions of the Atlantic barnacle Chthamalus proteus in the Pacific. Mol Ecol 14:3719–3733
Acknowledgments
This study would not have been possible without the generous help provided by various individuals. In particular we wish to thank Drs Ganesh Thiruchitranbalam (Marine Biological Laboratory, Dept of Zoology, Andhra University, India), Liu Li-lian (Institute of Marine Biology, National Sun Yat-Sen University, Kaohsiung, Taiwan), Chiu Yiuh-wen (Shu-Zen College of Medicine and Management, Kaohsiung, Taiwan) and Leung Kim-Fung (Environmental Protection Department, Hong Kong, China) for obtaining samples for analysis. We are also grateful to Dr Suzanne Williams (Department of Zoology, The Natural History Museum, London) for advice during the initial phase of this project, and to Dr Sujatha Kutty (Department of Biological Sciences, National University of Singapore) for her kind assistance in sequence analyses. Partial financial support provided by the Agency for Science, Technology and Research (A*STAR) to the Tropical Marine Science Institute, National University of Singapore through the Marine Environment Programme (2002–2004) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wong, Y.T., Meier, R. & Tan, K.S. High haplotype variability in established Asian populations of the invasive Caribbean bivalve Mytilopsis sallei (Dreissenidae). Biol Invasions 13, 341–348 (2011). https://doi.org/10.1007/s10530-010-9825-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-010-9825-5