
Abstract
Interactions between tumors and mesenchymal stem cells (MSCs) can regulate cancer cell behavior and cancer progression. Rat bone marrow-derived MSCs (rMSCs) were isolated and purified by Percoll density gradient centrifugation. Conditioned media from rMSCs (MSC-CM) was prepared, and its role in cancer cell migration and the underlying molecular mechanism were investigated. MSC-CM increased the migration and up-regulated the expression of CXC chemokine receptor 4 (CXCR4) in rat hepatoma cells (CBRH-7919). F-actin remodeling was observed, and the Young’s modulus was decreased in CBRH-7919 cells. A CXCR4 inhibitor suppressed the MSC-CM-induced CXCR4 expression and migration, restored the decrease in the Young’s modulus and disrupted the formation of F-actin. MSC-CM thus promotes CBRH-7919 cell migration by lessening cell stiffness and increasing F-actin formation through up-regulation of CXCR4 expression. MSC-CM may therefore have a positive impact on cancer metastases and underlines a potential safety issue associated with clinical applications of MSCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) are typically characterized by their ability to proliferate and give rise to other types of mesenchymal cells through differentiation, including osteoblasts, chondrocytes, adipocytes, muscle cells, pericytes and reticular fibroblasts, as well as neural cells in the ectoderm (Pittenger et al. 1999). These cells can be recruited to the sites of inflammation and injured tissues, which promotes their potential use as a repair kit for the healing of diseased and damaged tissues/organs and as tools for gene delivery and cell therapy. MSCs have become an attractive cell source and are widely employed in research and in the development of a variety of regenerative medicine and tissue engineering strategies (Caplan 2007).
Malignant tumors constitute a fatal disease to human health that are caused by a disorganized cell cycle and uncontrollable cell division and growth. Invasion and metastasis are the most insidious and life-threatening aspects of malignant tumors. At present, malignant tumors can be treated by surgery, chemotherapy, radiation therapy, immunotherapy, and monoclonal antibody therapy. However, these methods cannot achieve the goal of removing the cancer completely without damage to the rest of the body. Upon completion of therapy, the majority of cases present tumor regrowth and disease relapse (Palmer et al. 2011). Although marked progress has been obtained, the nosogenetic mechanism and cancer progression have not been fully clarified, and the ideal prevention and treatment methods have not yet been identified. Thus, the management of malignant tumor remains one of the most challenging worldwide problems in the clinic.
Inflammatory responses play decisive roles at different stages of tumor development, including initiation, promotion, malignant conversion, invasion, and metastasis. It has become evident that an inflammatory microenvironment is an essential component of all tumors (Grivennikov et al. 2010). In addition, the malignant change in normal tissue during tumorigenesis could also be considered a special type of tissue damage. Therefore, inflammatory mediators expressed by tumor tissue can recruit MSCs into the tumor microenvironment as part of the tumor remodeling process (Spaeth et al. 2008). The findings that MSCs can be recruited by tumors have triggered a series of studies to explore the potential effects of MSCs on cancer progression.
The relationship between MSCs and tumor cells is dual. MSCs recruited by tumors can remold the tumor microenvironment and affect the behaviors of tumor cells. When attracted by MSCs, tumor cells produce a variety of cytokines to affect MSC growth and survival (Bergfeld and DeClerck 2010). Many studies have reported the interactions between MSCs and tumor cells in primary tumors (Cousin et al. 2009; Prantl et al. 2010; Otsu et al. 2009; Li et al. 2010; Karnoub et al. 2007). Our previous study also demonstrated that conditioned medium from hepatocellular carcinoma cells promotes MSC migration (Qin et al. 2013). However, the crosstalk between MSCs and tumor cells has not yet been fully elucidated, and the precise molecular mechanisms involved in these processes remain unclear. In the present study, we aimed to determine the effects of MSC-CM on the migration of hepatoma cells and to explore the possible molecular mechanisms underlying the effects of MSC-CM on the migration of hepatoma cells.
Materials and methods
Cell isolation and cultivation
The rat hepatoma cell line (CBRH-7919) was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % (v/v) fetal bovine serum (FBS), 100 U penicillin/ml, and 100 μg streptomycin/ml in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. The culture medium was changed two to three times per week. Subculture was performed by digestion with 0.25 % trypsin/0.02 % EDTA when the cells were nearly confluent.
Rat MSCs were isolated and processed as previously described (Luo et al. 2009). Briefly, the femurs and tibias from two-month-old Sprague-Dawley rats (Laboratory Animal Center, the Third Military Medical University, China) were sawn, and the gelatinous bone marrow was extracted under sterile conditions. MSCs were obtained by density gradient centrifugation with 1.073 g Percoll/ml at 1,100×g for 30 min and then cultured in DMEM supplemented with 10 % (v/v) FBS, 100 U penicillin/ml and 100 μg streptomycin/ml in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. The culture medium was changed two to three times per week, and subculture was performed by digestion with 0.25 % trypsin/0.02 % EDTA when the cells were nearly confluent. Cells from passages 2–5 were used for the experiments.
Preparation of conditioned medium
For the preparation of the MSC-CM, the supernatant was collected after the MSCs were incubated for 24 h in serum-free DMEM (2 × 105 cells ml−1), centrifuged at ~1,100×g for 3 min and passed through a 0.22 μm filter. The MSC-CM was then stored at −4 °C until further use within one week.
Cell migration assay
The cell migration was assessed using 24-well Transwell chambers (pore size: 8 μm). Briefly, approx. 5 × 103 CBRH-7919 cells were harvested, suspended in 200 μl serum-free DMEM and placed in the upper chamber. The lower chambers contained 600 μl serum-free DMEM or MSC-CM. The plates were incubated at 37 °C in 5 % CO2 for 24 h, and the cells were then fixed in methanol for 15 min and stained with 0.05 % crystal violet in PBS for 20 min. The cells on the upper side of the filters were gently removed with cotton-tipped swabs, and the filters were washed with PBS. The cells on the underside of the filters were examined and counted under a microscope. Images were taken of three fields randomly selected from each insert. The number of cells in each field was counted and averaged. The cell migration is expressed as the fold increase compared with the corresponding control.
In the inhibition experiment, AMD3100 (Santa Cruz, CA, USA), a small molecule inhibitor of CXCR4, was dissolved in DMSO and stored at −20 °C. To inhibit the CXCR4 receptor, CBRH-7919 cells were incubated with AMD3100 (50 μg/ml) for 30 min at 37 °C before the cells were seeded into the culture insert.
Cell proliferation assay
The MTT assay was performed to evaluate the effect of MSC-CM on the proliferation of CBRH-7919 cells. CBRH-7919 cells were incubated with MTT (5 mg/ml) for 4 h, and formazan was dissolved by DMSO and quantified using a microplate reader. The relative proliferation rates were determined by normalizing the optical density value obtained for the experimental groups to that of the control group.
RNA extraction and reverse transcription PCR (RT-PCR)
The total RNA from CBRH-7919 cells was extracted using an RNA extraction kit (Qiagen). Reverse transcription was subsequently performed according to manufacturer’s instructions (RT reagent kit, Takara, Japan). The primers used for polymerase chain reaction (PCR) were designed using the Primer Premier 5.0 Software (Premie R Bio soft International, CA, USA) and synthesized by Invitrogen (Shanghai, China). The expression of the house-keeping gene β-actin was detected for normalization purposes. The primer pairs used in this study were as follows: β-actin(398bp): sense: 5′-CTGCCGCATCCTCTTCCTC-3′, antisense: 5′-CTCCTGCTTGCTGATCCACAT-3′; CXCR4(446bp): sense: 5′- GGGTTGGTAATCCTGGTC -3′, antisense: 5′-ATGATGTGCTGGAACTGG-3′. The reaction mixtures were incubated at 94 °C for 5 min, then at 94 °C for 30 s, 55.5 °C for 30 s and 72 °C for 30 s for 23 and 35 cycles for β-actin and CXCR4, respectively, and at 72 °C for 10 min. The amplified cDNA fragments were separated by electrophoresis on a 1.5 % agarose gel (Takara, Japan). The transcript expression of CXCR4 was determined using the Gel Doc XR Imaging System (Bio-Rad, CA, USA) and normalized to the expression of β-actin.
SDS-PAGE and western blot analysis
CBRH-7919 cells were treated with or without MSC-CM for 24 h. For the inhibition experiment, the cells were pretreated with AMD3100 (50 μg/ml) for 30 min and then treated with MSC-CM containing AMD3100 for another 24 h. Protein isolation was conducted using cell lysis buffer (62 mM Tris/HCl, pH 6.8, 10 % v/v glycerol, 2 % v/v SDS, 2 % v/v β-mercaptoethanol, and 0.02 % Bromophenol Blue) supplemented with a protease inhibitor cocktail (Roche, CA, USA) and phosphatase inhibitor cocktails I and II (Sigma-Aldrich). Following electrophoretic separation by 10 % (v/v) SDS-PAGE, the proteins were electroblotted onto PVDF membranes. The membranes were blocked with Tris-buffered saline containing 0.1 % Tween 20 (TBST) and 5 % (v/v) bovine serum albumin for 1 h at room temperature. Antibodies against CXCR4 and β-actin (Santa Cruz, CA, USA) were used according to the manufacturer’s protocol. Incubation was performed overnight at 4 °C with gentle shaking. Thereafter, the membranes were washed in TBST buffer, and a further incubation was performed with a HRP-conjugated antibody (goat anti-rabbit IgG, Sigma-Aldrich) for 1 h at room temperature. The protein expression was visualized using an enhanced electrogenerated chemiluminescence (ECL) system (Amersham, Chicago, IL, USA). The expression of CXCR4 at the protein level was normalized to that of the house-keeping protein β-actin.
Atomic force microscopy (AFM) analysis
CBRH-7919 cells were seeded on sterilized 12 mm coverslips and cultured in a six-well microplate in the incubator for two days. The cells were then exposed to MSC-CM with or without AMD3100 for 24 h. The Young’s elastic modulus of a single cell was measured using an atomic force microscope mounted on an inverted microscope at 37 °C. A soft silicon nitride quadratic pyramid tip (0.02 N/m) was used with an angle of 17.5°. A single cell with normal morphology was identified using the optical microscope, and the AFM cantilever probe was positioned on the perinuclear region. Each cell was mechanically probed with the AFM at three locations. The force curve was obtained by measuring the cantilever deflection at every vertical z-position of the cantilever as it approached and indented the cell. The cantilever was descended toward the cell at 2 μm/s until a trigger force of 2 nN was reached. The force-distance curves were collected and analyzed by JPK Imaging Process to obtain the Young’s elastic modulus (E).
Immunostaining
CBRH-7919 cells were prepared using a protocol similar to that adopted for the AFM analysis. The cells were then fixed with 2 % (v/v) paraformaldehyde in PBS for 15 min and permeabilized with 0.1 % Triton X-100 for 2 min. After saturation with 1 % bovine serum, the cells were incubated with phalloidin at 4 °C overnight. The nuclei were labeled with Hoechst 33258 for 5 min at room temperature. The cells were photographed with an inverted fluorescence microscope.
Statistical analysis
The data are represented as the mean ± standard deviation. The statistical analyses to compare the results between two groups were conducted using paired Student’s t test, and a value of p < 0.05 was considered statistically significant.
Results
MSC-CM promoted CBRH-7919 cell migration
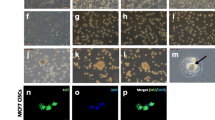
Tumor cell migration plays a crucial role in tumor procession. To determine whether MSC-CM affects the migration of CBRH-7919 cells, a transwell migration assay was employed to examine the change in CBRH-7919 cell migration in the presence of MSC-CM. As shown in Fig. 1, MSC-CM treatment significantly promoted the chemotaxis of CBRH-7919 cells compared with that of the control group.

MSC-CM promotes the migration of CBRH-7919 cells. a The migration assay was performed using a transwell system. MSC-CM was added to the lower chamber of the transwell system, and the transwell system was placed in the incubator. After 24 h, the migrated cells were stained with crystal violet (Scale bar represents 100 μm). b The migratory cell number was counted, and the migration is expressed as the change in the migratory rate relative to that of the control (designated 1.0). n = 3, **p < 0.01
To determine the proliferation of CBRH-7919 cells stimulated by MSC-CM, an MTT assay was performed. The results showed that MSC-CM had no obvious impact on the proliferation of CBRH-7919 cells within 24 h (Fig. 2) compared with the control. These results indicate that MSC-CM promotes the migration of CBRH-7919 cells without changing the cell number.

Effects of MSC-CM on the proliferation of rat hepatoma CBRH-7919 cells. The cells were seeded in 24-well plates and cultured overnight. After they were growth-arrested by serum-free DMEM for 24 h, MSC-CM was added, and the cells were cultured for an additional 24 h. The cell viability was determined by the MTT assay. The relative cell proliferation is expressed as the fold increase compared with the control (designated 100 %). n = 3
MSC-CM enhanced CXCR4 expression in CBRH-7919
Stromal cell-derived factor-1 (SDF-1) and its receptor, CXCR4, are crucial for the movement and migration of multiple cell types. We examined the effect of MSC-CM on CXCR4 expression in CBRH-7919 cells through RT-PCR and western blot analyses. As shown in Fig. 3, the presence of MSC-CM significantly enhanced the expression of CXCR4 in CBRH-7919 cells at both the mRNA (Fig. 3a) and protein levels (Fig. 3b).

Effects of MSC-CM on the expression of CXCR4 in CBRH-7919 cells. a The cells were incubated with MSC-CM for 24 h, and the effect of MSC-CM on the mRNA expression of CXCR4 was then detected by RT-PCR. The upper figure showed a representative gel electrophoresis result. The bottom figure shows the results of the densitometric and statistical analysis. The values represent the fold change in CXCR4 gene expression normalized to that of β-actin in response to MSC-CM stimulation relative to that found for the control cells (designated 1.0). n = 3, *p < 0.05. b The effect of MSC-CM on the protein expression of CXCR4 was detected by western blot. The upper figure shows a representative film image. The bottom figure shows the results of the densitometric and statistical analysis. The values represent the fold change in CXCR4 protein expression normalized to the β-actin level in response to MSC-CM stimulation relative to that found for the control cells (designed 1.0). n = 3, **p < 0.01
CXCR4 up-regulation was involved in MSC-CM-induced CBRH-7919 cell migration
Because we demonstrated that MSC-CM up-regulates CXCR4 expression in CBRH-7919 cells, we hypothesized that the up-regulation of CXCR4 expression induced by MSC-CM may play an important role in MSC-CM-enhanced CBRH-7919 cell migration. To test this hypothesis, CBRH-7919 cells were pretreated with the CXCR4 inhibitor AMD3100 for 30 min before being co-cultured with MSC-CM. As shown in Fig. 4, in the presence of AMD3100, the increased expression of CXCR4 induced by MSC-CM in CBRH-7919 cells was inhibited at the mRNA (Fig. 4a) and protein levels (Fig. 4b). Moreover, the presence of AMD3100 also abolished the MSC-CM-promoted migration of CBRH-7919 cells (Fig. 4c), indicating that the up-regulation of CXCR4 induced by MSC-CM is involved in the MSC-CM-promoted migration of CBRH-7919 cells.


Effects of the CXCR4 inhibitor AMD3100 on MSC-CM-induced migration and the expression of CXCR4 in CBRH-7919 cells. The cells were incubated with AMD3100 (50 μg/ml) for 30 min at 37 °C and were then seeded into the culture insert. AMD3100 inhibited the MSC-CM-induced expression of CXCR4 at both the mRNA (a) and protein levels (b). n = 4, *p < 0.05, **p < 0.01. AMD3100 also suppressed the MSC-CM-induced migration of CBRH-7919 cells (c). n = 3, *p < 0.05, **p < 0.01
MSC-CM reduced the stiffness of CBRH-7919 cells via CXCR4 pathways
The cell migratory ability is closely related to cell stiffness, which is characterized by the Young’s modulus. Based on the observation that MSC-CM promoted the migration of CBRH-7919 cells, we used an atom force microscope to examine the effect of MSC-CM on the Young’s modulus of CBRH-7919 cells and found that the Young’s modulus of the MSC-CM-treated cells was significantly reduced compared with that of the control cells. However, in the presence of the CXCR4 inhibitor AMD3100, the MSC-CM-induced decrease in the Young’s modulus of CBRH-7919 cells was restored (Fig. 5). This result suggested that MSC-CM can reduce the stiffness of CBRH-7919 cells via CXCR4 pathways.

Atomic force microscopy (AFM) analysis revealed differences in the cell stiffness, as represented by the Young’s modulus. CBRH-7919 cells exposed to MSC-CM were less stiff (lower Young’s modulus) than the control, whereas the CXCR4 inhibitor AMD3100 prevented the decrease in the Young’s modulus induced by MSC-CM. The data are expressed as the mean ± SD. n = 9, *p < 0.05
MSC-CM remodeled F-actin in CBRH-7919 cells
Because cell stiffness is usually affected most strongly by the cytoskeleton, we then employed confocal microscopy and examined the effect of MSC-CM on the F-actin cytoskeleton in CBRH-7919 cells. As shown in Fig. 6, MSC-CM significantly remodeled the cytoskeleton and enhanced the fluorescence intensity of F-actin in CBRH-7919 cells, whereas AMD 3100 inhibited the remodeling of F-actin induced by MSC-CM, indicating that MSC-CM can influence F-actin remodeling through CXCR4 signaling in CBRH-7919 cells.

Treatment with MSC-CM regulated actin cytoskeleton remodeling and expression. a Immunofluoresence staining of F-actin in control cells, CBRH-7919 cells exposed to MSC-CM for 24 h, and CBRH-7919 cells exposed to MSC-CM + AMD3100 (50 μg/ml) for 24 h. The inserts in the figures present the details at an enlarged scale. Bars = 100 μm. b The representative bands show the average optical density of F-actin (E). For each group, 17 cells were randomly chosen from three independent experiments, and the fluorescence intensity of F-actin was analyzed. The data are expressed as the mean ± SD; n = 17; **p < 0.01
Discussion
MSCs are attractive candidates for cell-based tissue repair because of their self-renewal ability and their multi-lineage differentiation potential. They are also used as vectors for delivering therapeutic genes to sites of injury due to their migratory behavior (Dimarino et al. 2013). An increasing number of studies are confirming the promising application of MSCs for the repair and regeneration of damaged tissue in the clinic. However, MSCs can be mobilized and recruited to tumor tissue sites where they can be incorporated into the tumor’s microenvironment and potentiate further tumorigenesis and/or tumor progression via differentiation into tumor-nurturing stroma, interaction with cancer cells by direct cell contact or the release of paracrine factors (Zimmerlin et al. 2013; Spaeth et al. 2008; Roorda et al. 2010; Spaeth et al. 2009). Many components of MSCs that are required for tissue repair and regenerative therapy are also critical for tumor progression and metastasis (Cuiffo and Karnoub 2012). Hence, there has been a growing interest in the study of the role of MSCs in cancer progression over the last decade.
A bidirectional crosstalk between tumor cells and MSCs has been demonstrated in a variety of cancers (Bergfeld and DeClerck 2010). Moreover, some studies have suggested that the influence of MSCs on cancer progression can be either an anti- or a pro-tumor effect. MSCs derived from adipose tissue inhibit pancreatic cancer proliferation both in vitro and in vivo and induce tumor cell death by altering cell cycle progression (Cousin et al. 2009), whereas adipose tissue-derived mesenchymal stem cells subcutaneously co-injected with prostate cancer cells promote prostate tumor growth (Prantl et al. 2010). Human MSCs (hMSCs) inhibit the malignant phenotypes of human liver cancer cell lines H7402 and HepG2 both in vitro and in vivo, including their proliferation, colony-forming ability and oncogene expression (Qiao et al. 2008). However, our previous study showed that rMSCs significantly enhance the proliferation of the rat hepatoma cell line CBRH-7919 (unpublished data). Unexpectedly, opposite proliferation effects on the same type of tumor cells have also been reported in studies of MSCs from different sources. For example, the subcutaneous injection of rMSCs into a melanoma rat model led to an inhibitory effect on the growth of tumor tissue (Otsu et al. 2009). In contrast, mouse MSCs co-injected subcutaneously with melanoma cells favor tumor growth (Djouad et al. 2003). In addition, hMSCs inhibit the metastasis of hepatocellular carcinoma MHCC97 cells (Li et al. 2010). However, in the present study, we found that conditioned medium from rMSCs promoted the migration of rat hepatoma CBRH-7919 cells, which is consistent with the findings of a previous study showing that MSCs within tumor stroma promote breast cancer metastasis (Karnoub et al. 2007). These results reinforce that the effects of local or systemic MSCs on cancer progression remain controversial and that the factors involved in this process are very complex (Klopp et al. 2011; Wong 2011). The effects may vary widely due to differences in the source of MSCs and the tumor model.
Although MSCs have received much attention in the field of stem cell transplantation because of their promising use as tools for the healing of diseased and damaged tissues and for gene delivery and cell therapy, the fact that the effects of MSCs on cancer progression are controversial also warns that the security issue associated with the use of MSCs in clinical applications cannot be ignored. It has been reported that MSC transplantations may result in some dangerous disadvantages, such as teratomas formation by MSCs (Herberts et al. 2011). To avoid this possible safety concern, recent attention hasbeen focused on using MSC-CM to develop a cell-free therapeutic approach in stem cell therapy (Timmers et al. 2011; Ivanova-Todorova et al. 2012; Osugi et al. 2012; Cantinieaux et al. 2013). However, Zhang et al. (2013) found that both co-culturing with MSCs and treatment with MSC-CM enhances the growth of 4T1 breast cancer cells. Our data also show a significantly increased migratory ability of rat hematoma CBRH-7919 cells in the presence of MSC-CM. These results further demonstrate the risk of promoting growth and metastasis by MSCs or cell-free MSC-CM, indicating a safety concern with stem cell therapies after cancer.
As mentioned above, the effects of MSCs on tumor cells are multiple and controversial, which make the interpretation of their role in cancer progression more complex. The paradoxical effect of MSCs on cancer progression is currently poorly understood due to the limited studies examining the mechanisms underlying the interactions between MSCs and tumor cells. SDF-1, also known as C-X-C motif chemokine 12 (CXCL12), and its receptor, CXCR4, are chemokine proteins. The binding of SDF-1 to CXCR4 initiates divergent signaling pathways and is crucial for the homing and migration of multiple cell types in physiological or pathological settings (Molyneaux et al. 2003). MSCs secrete a large variety of molecules; thus, the complexity and diversity of the MSC-CM make it difficult to identify every relevant component (Timmers et al. 2011). MSCs can also secrete SDF-1 (Zhang et al. 2007; Landry et al. 2010), which drove us to examine the possible role of the SDF-1/CXCR4 axis in the MSC-CM-promoted migration of CBRH-7919 cells.
We found that the MSC-CM up-regulated the expression of CXCR4 in CBRH-7919 cells. Moreover, AMD3100, an inhibitor of CXCR4, not only suppressed the MSC-CM-induced expression of CXCR4 but also abolished the MSC-CM-promoted migration of CBRH-7919 cells. Thus, our results confirm that the up-regulation of CXCR4 by MSC-CM is involved in the MSC-CM-promoted migration of CBRH-7919 cells. In our study, AMD3100 decreased the MSC-CM-induced increase in CXCR4 expression in CBRH-7919 cells at both the mRNA and protein levels, suggesting that AMD3100 affects the expression of CXCR4 at the transcriptional level. Moreover, the down-regulation of CXCR4 expression may also result from translational modulation by AMD3100 because it has been suggested that miR-146a is modulated by AMD3100 and affects the expression of CXCR4 in leukemic blast cells (Spinello et al. 2011). However, the confirmation of existence of the latter mechanism in the model used in this study requires further work.
The cell migration ability is closely related to the cell stiffness (Oh et al. 2012). Malignant human breast epithelial cells are less stiff than non-malignant cells which allows cancerous cells to more easily migrate through the surrounding tissue matrix and small capillaries (Li et al. 2008). Our results show that the MSC-CM lessened the stiffness of CBRH-7919 cells. The SDF-1/CXCR4 axis, which is involved in the MSC-CM-promoted migration of CBRH-7919 cells, also plays an important role in the change in cell stiffness induced by MSC-CM.
These data suggest that the promotion of CBRH-7919 cell migration may be attributed to the decrease in cell stiffness stimulated by the MSC-CM. The cell stiffness is determined by the cytoskeleton, an intracellular polymeric network consisting of actin filaments, microtubules, intermediated filaments and nuclear skeleton proteins. Generally, a well-organized actin structure may stiffen cells. Interestingly, we found that an increase in F-actin formation is accompanied with a lessening in cell stiffness. Our previous study showed that the mechano growth factor promotes rat tenocyte migration by lessening cell stiffness and increasing F-actin formation (Zhang et al. 2014). Based on these findings, we speculate that MSC-CM may lessen the nuclear stiffness, which plays an important role in determining the superimposed Young’s modulus of CBRH-7919 cells. However, more research is needed to confirm this in our next study.
In summary, we have investigated changes in CBRH-7919 cell migration and the underlying molecular mechanism in the presence of MSC-CM and demonstrated that MSC-CM can promote the migration of rat hepatoma cells through CXCR4 up-regulation and F-actin remodeling. Our studies also provide evidence that even the cell-free MSC-CM may have a positive impact on metastases. Such results warrant careful monitoring of MSCs use in patients diagnosed with cancer.
References
Bergfeld SA, DeClerck YA (2010) Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev 29:249–261
Cantinieaux D, Quertainmont R, Blacher S, Rossi L, Wanet T, Noël A, Brook G, Schoenen J, Franzen R (2013) Conditioned medium from bone marrow-derived mesenchymal stem cells improves recovery after spinal cord injury in rats: an original strategy to avoid cell transplantation. PLoS One 8:e69515
Caplan AI (2007) Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J Cell Physiol 213:341–347
Cousin B, Ravet E, Poglio S, De Toni F, Bertuzzi M, Lulka H, Touil I, André M, Grolleau JL, Péron JM, Chavoin JP, Bourin P, Pénicaud L, Casteilla L, Buscail L, Cordelier P (2009) Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS One 4:e6278
Cuiffo BG, Karnoub AE (2012) Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell Adh Migr 6:220–230
Dimarino AM, Caplan AI, Bonfield TL (2013) Mesenchymal stem cells in tissue repair. Front Immunol 4:201
Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, Noel D, Jorgensen C (2003) Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood 102:3837–3844
Grivennikov SI, Greten FR, Karin M (2010) Immunity, Inflammation, and Cancer. Cell 140:883–899
Herberts CA, Kwa MS, Hermsen HP (2011) Risk factors in the development of stem cell therapy. J Transl Med 9:29
Ivanova-Todorova E, Bochev I, Dimitrov R, Belemezova K, Mourdjeva M, Kyurkchiev S, Kinov P, Altankova I, Kyurkchiev D (2012) Conditioned medium from adipose tissue-derived mesenchymal stem cells induces CD4+FOXP3+ cells and increases IL-10 secretion. J Biomed Biotechnol 2012:295167
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA (2007) Mesenchymal stem cells within tumor stroma promote breast cancer metastasis. Nature 449:557–563
Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F (2011) Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 29:11–19
Landry Y, Le O, Mace KA, Restivo TE, Beauséjour CM (2010) Secretion of SDF-1 alpha by bone marrow-derived stromal cells enhances skin wound healing of C57BL/6 mice exposed to ionizing radiation. J Cell Mol Med 14:1594–1604
Li QS, Lee GY, Ong CN, Lim CT (2008) AFM indentation study of breast cancer cells. Biochem Biophys Res Commun 374(4):609–613
Li GC, Ye QH, Xue YH, Sun HJ, Zhou HJ, Ren N, Jia HL, Shi J, Wu JC, Dai C, Dong QZ, Qin LX (2010) Human mesenchymal stem cells inhibit metastasis of a hepatocellular carcinoma model using the MHCC97-H cell line. Cancer Sci 101:2546–2553
Luo Q, Song G, Song Y, Xu B, Qin J, Shi Y (2009) Indirect coculture with tenocyte promotes the proliferation and mRNA expression of tendon/ligament related genes in mesenchymal stem cells. Cytotechnology 61:1–10
Molyneaux KA, Zinszner H, Kunwar PS, Schaible K, Stebler J, Sunshine MJ, O’Brien W, Raz E, Littman D, Wylie C, Lehmann R (2003) The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development 130:4279–4286
Oh MJ, Kuhr F, Byfield F, Levitan I (2012) Micropipette aspiration of substrate-attached cells to estimate cell stiffness. J Vis Exp 67:3886
Osugi M, Katagiri W, Yoshimi R, Inukai T, Hibi H, Ueda M (2012) Conditioned media from mesenchymal stem cells enhanced bone regeneration in rat calvarial bone defects. Tissue Eng Part A 18:1479–1489
Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J (2009) Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood 113:4197–4205
Palmer TD, Ashby WJ, Lewis JD, Zijlstra A (2011) Targeting tumor cell motility to prevent metastasis. Adv Drug Deliv Rev 63:568–581
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Prantl L, Muehlberg F, Navone NM, Song YH, Vykoukal J, Logothetis CJ, Alt EU (2010) Adipose tissue-derived stem cells promote prostate tumor growth. Prostate 70:1709–1715
Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC, Ye L, Zhang X (2008) Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res 18:500–507
Qin X, Luo Q, Zhang BY, Shi YS, Song GB (2013) Conditioned medium from hepatocellular carcinoma cells promotes mesenchymal stem cells migration through CXCR4-ERK1/2 signal pathway. J Biol Res-Thessalon 20:259–269
Roorda BD, At Elst, Boer TG, Kamps WA, de Bont ES (2010) Mesenchymal stem cells contribute to tumor cell proliferation by direct cell-cell contact interactions. Cancer Invest 28:526–534
Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F (2008) Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther 15:730–738
Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F (2009) Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One 4:e4992
Spinello I, Quaranta M, Riccioni R et al (2011) MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J 1:e26
Timmers Lim SK, Hoefer IE, Arslan F, Lai RC, van Oorschot AA, Goumans MJ, Strijder C, Sze SK, Choo A, Piek JJ, Doevendans PA, Pasterkamp G, de Kleijn DP (2011) Human mesenchymal stem cell-conditioned medium improves cardiac function following myocardial infarction. Stem Cell Res 6:206–214
Wong RS (2011) Mesenchymal stem cells: angels or demons? J Biomed Biotechnol 2011:459510
Zhang M, Mal N, Kiedrowski M, Chacko M, Askari AT, Popovic ZB, Koc ON, Penn MS (2007) SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes aftermyocardial infarction. FASEB J 21:3197–3207
Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G (2013) Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Ther 4:70
Zhang B, Luo Q, Mao X, Xu B, Yang L, Ju Y, Song G (2014) A synthetic mechano-growth factor E peptide promotes rat tenocyte migration by lessening cell stiffness and increasing F-actin formation via the FAK-ERK1/2 signaling pathway. Exp Cell Res 322:208–216
Zimmerlin L, Park TS, Zambidis ET, Donnenberg VS, Donnenberg AD (2013) Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie 95:2235–2245
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 11032012, 11102240, and 11272365), the 111 Project (No. 06023), the Visiting Scholar Foundation of Key Laboratory of Biorheological Science and Technology (Chongqing University), the Ministry of Education of China (No. CQKLBST-2012-008), and the Research Fund for the Doctoral Program of Higher Education of China (No. 20130191110029).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, X., Luo, Q., Sun, J. et al. Conditioned medium from mesenchymal stem cells enhances the migration of hepatoma cells through CXCR4 up-regulation and F-actin remodeling. Biotechnol Lett 37, 511–521 (2015). https://doi.org/10.1007/s10529-014-1710-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-014-1710-3