
Abstract
A bacterium with lipolytic activity was isolated from the Chukchi Sea within the Arctic Ocean. The lipase BpL5 from the isolate, Bacillus pumilus ArcL5, belongs to subfamily 4 of lipase family I. The optimum pH and temperature of the recombinant enzyme BpL5, as expressed in Escherichia coli, were 9.0 and 20 °C, respectively. The enzyme retained 85 % of its activity at 5 °C. There was a significant difference between temperatures for maximal activity (20 °C) and for protein denaturation (approx. 45 °C). The enzyme preferred middle-chain (C8) p-nitrophenyl substrates. Two mutants, S139A and S139Y, were rationally designed based on the 3D-structure model, and their activities were compared with that of the wild type. The both mutants showed significantly improved activity against tricaprylin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipases (EC. 3.1.1.3) are produced by microorganisms, animals, and plants. They have attracted much interest for biotechnological applications because their catalytic activities are related to hydrolysis and synthesis reactions with high regioselectivity and enantioselectivity (Gupta et al. 2004). Their applications are mainly in the detergent, food, agrochemicals, and pharmaceutical industries as well as being used to produce new biopolymeric materials (Joseph et al. 2007; Schmid and Verger 1998).
Bacterial lipases are classified into eight families. Family I, the largest family, contains six subfamilies. Bacillus lipases, that display marked biotechnological potentials because of their selectivity toward racemic substrates (Detry et al. 2006; Rasool et al. 2005), belong to subfamilies four and five, in which the first glycine residue in the conserved G-X-S-X-G pentapeptide around the active-site serine residue is replaced by alanine. The bacterial origins of subfamily 4 lipases have been reported only for a few Bacillus species, viz., Bacillus subtilis, Bacillus licheniformis and bacillus pumilus.
Psychrophilic bacteria that live at low temperatures have colonized all permanently cold environments, from the deep sea to the polar regions. This requires physiological and biochemical adaptations. As enzymes tend to become rigid at low temperatures, psychrophilic enzymes must evolve to improve their dynamics or their flexibility to achieve catalysis. These structural features of psychrophilic enzymes can compensate for the exponential decrease of chemical reaction rates induced by lowered temperatures. These characteristics of psychrophilic enzymes make them useful as additives to detergents, as biocatalysts for biotransformation of labile compounds at cold temperatures, and for bioremediation of polluted soils and wastewaters at low temperatures (Joseph et al. 2007, 2008).
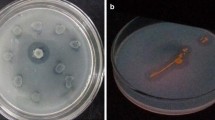
Psychrophilic lipases are widely distributed in bacteria and yeasts living at low temperatures (~5 °C) (Joseph et al. 2007). We have previously isolated psychrophilic bacteria from arctic seawater, some which demonstrate lipase activity at low temperatures. Using screening on an agar plate containing tricaprylin (TCN), the bacterial strain with highest lipolytic activity was identified as B. pumilis.
In this study, we have cloned the lipase gene from B. pumilus and characterized the recombinant enzyme after expression in Escherichia coli. Furthermore, two rational mutants of the lipase were designed based on functional prediction from the structural model, and the enzymatic activity of these mutant also characterized.
Materials and methods
Bacterial strain
Escherichia coli strains DH5α and BL21(DE3)pLysS was used for the cloning and expression steps, respectively.
Gene cloning and sequence analysis
Oligonucleotide primers for PCR were purchased from Macrogen (Seoul). DNA extraction and purification kits were from Qiagen (Germany), KOD DNA polymerase (Toyobo). Restriction enzymes were from New England Biolabs. All of DNA manipulations were performed by standard methods. The genomic DNA was prepared for PCR by DNA purification kit. The lipase gene was amplified using primers: Bplip1-f (5-GAGTCGATAAGATGAATAAGGGGGAATG-3) with Bplip1-r (TTAATTCGTATTTTGTCCTCCGCCGTTC), which were designed according to the sequence of the previous B. pumilus lipases (Bell et al. 2002).
Expression of the lipase gene in E. coli
The expression vector pET22b and the host strain BL21(DE3)pLysS was purchased from Novagen. To construct the expression plasmid vector, two types of primer sets were designed. One set is for the expression of the full-length lipase gene containing signal peptide. The other is for the mature form without signal sequence. The former primer set is ArcL5-f (5-AAGAAGGAGATATACATATGAAAGTGATTTGTTTTAAGA-3) and ArcL5-r (5-GTGGTGGTGCTCGAGATTCGTATTTTGTCCTCCGC-3). The latter primer set is ArcL5-mf (5-AAGAAGGAGATATACATATGGCTGAGCACAATCCGGTTG-3) and ArcL5-r. The ORF was amplified directly from the genomic DNA of the ArcL5 strain. The vector pET22b and the both PCR products encoding the full-length gene and the mature gene, respectively, were ligated and the expression vectors were constructed using the ligation-independent cloning method (Jeong et al. 2012), respectively. E. coli BL21(DE3)pLysS transformed with the expression vector was grown overnight with shaking (180 rpm) at 37 °C in 10 ml LB broth and ampicillin (0.01 %). The inoculum was transferred into 1 l LB broth with ampicillin. Induction of the recombinant proteins was performed by the following protocol. When the OD600 reached 0.6, IPTG was added at 0.1 mM, and the temperature was lowered to 25 °C. After overnight of cultivation, cells were harvested by centrifugation at 3,200×g at 4 °C for 20 min, and frozen at −80 °C until the next experiment.
Purification of the recombinant proteins
Soluble proteins were recovered from the cell extract of the recombinant containing pBpL5 m that was induced by IPTG as described the above. All of the recombinant proteins were fused with 6×His-tag peptide at the C-terminal region. Purification of the recombinant proteins was carried out by Ni-affinity chromatography on a HisTrap HP column. The soluble proteins was loaded on Ni-affinity column, which had already been equilibrated with buffer A (20 mM Tris/HCl, pH 8.0, 500 mM NaCl and 10 mM imidazole). The target protein was eluted with buffer B (20 mM Tris/HCl, pH8.0, 500 mM NaCl and 400 mM imidazole). Fractions with the lipase activity were combined and were concentrated by Vivaspin centrifugal filter devices MWCO 10,000 Da (Sartorius). The concentrated sample was applied on a gel filtration column packed with Superdex 75 preparation grade. Protein purity was assessed using SDS-PAGE analysis. Protein concentration was determined from the absorbance at 280 nm, using 15,930 M−1 cm−1 as the molar extinction coefficient.
Enzymatic activity assay
The general lipase activity was measured by spectrometrically using p-nitrophenyl caprylate (PNP-C8). The reaction mixture was consisted of 100 μM PNP-substrate, 4 % ethanol, 20 mM Tris/HCl buffer (pH 8.0), and an appropriate amount of the enzyme solution. One unit of the activity was defined as the amount of enzyme releasing 1 μmol p-nitrophenol per min under the assay conditions. The appreciate calibration curves were performed in the same buffer and temperature to calculate the corresponding molar absorptivities at 410 nm. Lipase activity against various triacylglycerides [TCN, olive oil, soybean, castor oil, sunflower, and palm oil] was performed by assaying the free fatty acids released by hydrolysis of the substrate with pH-stat instrument by titration with 10 mM NaOH. The amount of enzyme catalyzing the release of 1 μmol fatty acid per min was defined as 1 unit.
Molecular modeling
The 3D structure model of the BpL5 from B. pumilus ArcL5 was predicted using the I-TASSER online server (Zhang 2008) that generates a 3D structure model along with its confidence score (C-score) using a PDB database. Structural quality was analyzed using PROCHECK (Laskowski et al. 1996). Ribbon and stick representations were prepared with PyMOL (DeLano Scientific).
Construction of the mutants
Point mutation were performed using site-directed mutagenesis following the Quick-Change mutagenesis method (Stratagene). The following primer pairs were used to create the mutants: the primers S139A-f (5-ATA TGA TTG TTG TCA ACG CTC TCT CGC GTT-3) and S139A-r (5-AAA CGC GAG AGA GCG TTG ACA ACA ATC ATA-3) for S139A mutant, S139Y-f (5-ATA TGA TTG TTG TCA ACT ACC TCT CGC GTT-3) and S139Y-r (5-AAA CGC GAG AGG TAG TTG ACA ACA ATC ATA-3) for S139Y mutant. The mutant enzymes were expressed and purified according to the above method.
Circular dichroism (CD) spectroscopy
Experiments were conducted by monitoring protein denaturation of the wild type and its mutants in a Chirascan circular dichroism spectropolarimeter (Applied Photophysics, UK). Thermal denaturation transitions were followed by stepwise monitoring ellipticity changes at 220 nm with a 1 mm pathlength cuvette while the temperature was ramped from 20 to 70 °C at intervals of 2 °C. Plots were produced by defining the upper and lower temperature baselines as 0 and 100 %, respectively. The denaturation temperatures (Tm1/2) were decided as the point at which 50 % of the sample denatured. Scans were performed in duplicate and averaged.
Results and discussion
Isolation and identification of psychrophilic bacterium possessing lipolytic activity
A sample of seawater was collected from the Chuckchi Sea (N 75.31.24 W 178.47.04) in the arctic. To isolate psychrophilic bacteria with lipolytic activity, colonies with a clear halo zone were selected from agar plates of marine medium containing TCN, at 15 °C. Lipase activity of the isolates was confirmed by the enzyme assay method using the culture broth. Among them, a bacterial strain with the highest activity was identified as B. pumilis based on 16S DNA sequence analysis and named B. pumilis ArcL5.
Lipases from B. pumilus are classified as members of subfamily 4 and have sequence identities of more than 74 %. Subfamily 4 lipases originate mainly from Bacillus species (B. subtilis, B. pumilus, and B. licheniformis) (Jaeger and Eggert 2002). While these bacteria are known as mesophilic strains, strain ArcL5 grew well at or below 15 °C.
Cloning and sequence analysis of the lipase gene
The lipase gene from the strain ArcL5, named BpL5 (GenBank accession number: KF939143), was cloned using the specific primer for B. pumilus lipase (Bell et al. 2002). The ORF encoded a protein of about 19 kDa, which had the highest sequence identity (94 %) with the lipase of B. stratosphericus and also had high similarity to other B. pumilus lipases. The mature form of the lipase showed 81 % identity with that from B. subtilis and 65 % with that from B. licheniformis.
The pentapeptide (A–X-S-X-G), conserved in subfamily 4, was present as the sequence A-H–S-M-G, in the ORF of BpL5 (Fig. 1). Although the mature peptide of the lipase has very high similarity with other Bacillus lipases, the signal peptide sequences showed very low identity values with other Bacillus lipase signal peptide sequences.

Alignment of the amino acid sequence of the lipase BpL5 from B. pumilus ArcL5 strain with sequences of the subfamily 4 lipases from other Bacillus species. CLUSTAL W program was used for alignment. B. pumilus (the BpL5 protein), B. subtilis (PDB ID: 1i6w), B. amyloliquefaciens (GenBank ID: gi452854308), B. licheniformis (GenBank ID: gi511063369). Identical residues are shown as dots. The catalytic triad residues in the active site were marked by red colored circles. The conserved pentapeptide (A–X-S-X-G) was underlined in red. Residue for the mutation was marked by red arrow and boxing
Expression and characterization of the psychrophilic lipase
The full-length coding sequence of the gene, containing the sequence coding for the signal peptide, was amplified from the genomic DNA of strain ArcL5 using specific primers and then the PCR gene product was cloned into an expression plasmid. The resulting recombinant plasmid, pBpL5, was introduced into an expression host. Another recombinant plasmid, pBpL5m was also constructed for production of the mature form of the enzyme, but lacking the signal peptide sequence. As shown in Fig. 2, while the soluble and catalytically-active form was expressed in E. coli BL21(DE3)pLysS containing pBpL5m. Cells containing pBpL5, which retained the signal peptide sequence, exhibited little lipolytic activity and their growth rate was also markedly lower than that of cells harbouring pBpL5m.

SDS-PAGE analysis of the BpL5 enzyme expressed in E. coli BL21(DE3)pLysS. IPTG induction and purification methods were described in the section “Materials and methods”. Lane 1, the cell pellet; lane 2, the culture supernatant; lane 3, Insoluble fraction of the cell lysate; lane 4, Soluble fraction of the cell lysate. The purified enzyme, BpL5 was shown on the right panel
The enzyme was purified using Ni-affinity and gel filtration. According to SDS-PAGE analysis, the molecular weight of the purified BpL5 was 19 kDa, which is in agreement with the molecular mass (19,257 Da) calculated from the gene sequence.
To examine substrate specificity, we tested various p-nitrophenyl (PNP) fatty acyl esters (C4, C8, and C12) under at pH 8.0 and 25 °C. Kinetic parameters of the enzymes were calculated from progress curve analyses of PNP-substrate hydrolysis using the software ENCORA 1.2 (Straathof 2001; Willeman et al. 2000). The purified BpL5 exhibited an about 13-fold lower Km value for PNP-C8 compared to PNP-C4 and PNP-C12 (Table 1). Among the PNP-esters tested, the catalytic efficiency of the enzyme toward PNP-C8 exhibited the highest value. PNP-C8 was, therefore, used as the substrate to investigate the biochemical properties of the enzyme. The enzyme exhibited a sharp optimal activity at around pH 9, but with lower activity in glycine/NaOH buffer compared to that in Tris/HCl buffer at the same pH (Fig. 3).

Effects of pHs on the lipase activity. Enzyme activity was assayed at different pHs (black circle Na/acetate (pH 4.0–5.6), black square PBS (pH 5.2–7.45), black rhombus Tris/HCl (pH 8.0–9.0), and × glycine/NaOH (pH 9.0–10.0) using PNP-C8 as substrate. The assay method is described in the section “Materials and methods”. 100 % activity values correspond to specific activities of 954 units/μg of protein
The optimum temperature for the lipase activity was about 20 °C (Fig. 4) but even at 5 °C it showed about 85 % of its maximum activity. This optimization of the enzyme activity at low temperature was an expected result, given the habitat temperature of strain ArcL5, the origin of BpL5 (Fig. 4). This clear psychrophilic feature has not yet been observed in other B. pumilus lipases. B. pumilus lipases previously isolated from the Antarctic have had optimal temperatures of 40 °C (Arifin et al. 2013). To investigate the relationship between the activity and the stability of the enzyme, the thermal denaturation of the lipase was followed by continuous monitoring of the ellipticity at 220 nm during constant heating. The temperature at the middle of the denaturation transition (Tm1/2) of BpL5 was recorded as about 45 °C (Fig. 4).

Effects of temperatures on the activity and the thermal denaturation curves of the wild type and its mutants (black circle WT, white circle S139A, and downward triangle S139Y). Relative activity was calculated with value of the wild type at 20 °C, 100 % as basis value. Thermal denaturation of proteins was monitored at pH 8.0 by far UV circular dichroism (220 nm). Ellipticities were converted to the apparent denatured fraction. Detailed experiment was described in the section “Materials and methods”
These results suggested that this enzyme was inactivated at a temperature below that at which the protein was denatured. Generally, maximum enzymatic activity of mesophilic and thermophilic enzymes tends to be best at their denaturation temperature. However, in the case of BpL5, the temperatures for activity and stability did not match. The active site of psychrophilic enzyme (or the catalytic intermediates) is more heat-labile than the enzyme structure as a whole, because of its flexibility. Thus, this “localized flexibility in the active site” is a representative feature of psychrophilic enzymes (D’Amico et al. 2003; Feller and Gerday 2003; Fields and Somero 1998).
Protein structure prediction and mutational analysis
A structural model for BpL5 was predicted using the I-TASSER server (Zhang 2008), and exhibited a structure highly similar to that of B. subtilis lipase (PDB ID: 1i6w) (Fig. 5). As shown in the alignment presented in Fig. 1, the sequence similarity extends along all of the protein sequence. The crystal structure of B. subtilis lipase was shown as a compact minimal α/β hydrolase fold that lacks structural elements of the canonical α/β-hydrolase fold and without a lid domain covering the active site (van Pouderoyen et al. 2001). The active site of the structural model is solvent-exposed. The active cleft was relatively wide and shallow.

Comparison of the structure model of the BpL5 (yellow) and the crystal structure of B. subtilis lipase (cyan). The catalytic triad residues that consist of Ser77, Asp133, and His156 are represented as stick models. In the right panel, the structural position of the designed mutation was shown as stick model, together with the corresponding residue of B. subtilis lipase, Tyr139
The enzyme–inhibitor complexed model from B. subtilis lipase structure has suggested some substrate-binding patterns in the active cleft (van Pouderoyen et al. 2001). Using this information, residue Ser139 was selected as one of the predicted functional residues related to the substrate binding mechanism, which is not conserved in subfamily 4 (Fig. 1). Hence, two mutants, S139A and S139Y, were prepared for functional analysis. The optimal temperature and pH of the enzyme were not altered by these mutations.
The structural integrity of the mutants was checked by measuring their CD spectra. As shown in Fig. 6, while the spectral pattern of the S139A mutant was similar to that of the wild-type, the spectral pattern of the S139Y mutant showed a large shift from the pattern of the wild-type. This result suggested that the structure of the S139Y mutant is partially disrupted by the mutation.

Secondary structures of the wild type and its mutants. Secondary structures were predicted by scanning 190–260 nm ten-times and scans were subtracted by base line and averaged
When monitoring protein denaturation, the S139A mutant showed the same denaturation temperature (45 °C) as the wild type, but the S139Y mutant revealed a slightly decreased thermal stability, with a Tm½ value of 40 °C (Fig. 4). This decrease in stability may be caused by its partially disrupted structure, even though the substituted residue is exposed at the surface in the structural model. However, the activity of the S139Y mutant with respect to TCN was higher than that of the wild type. In addition, the S139A mutant demonstrated significantly improved activity against the two lipase substrates: PNP-substrates and triacylglycerols (Table 2).
In conclusion, we have isolated the lipase BpL5, possessing a psychrophilic character, from an Arctic Sea microorganism. To improve its activity, we have designed and engineering two mutants by exchanging an amino acid residue that is located nearby a putative substrate-binding cleft. This mutational approach caused a marked difference in the lipolytic activity of the BpL5 enzyme, showing that rational design may help to find regions important for enzyme activity. Consequently, the knowledge obtained through this study provides a biotechnological basis for creating an improved lipase for industrial application.
References
Arifin AR, Kim SJ, Yim JH, Suwanto A, Kim HK (2013) Isolation and biochemical characterization of Bacillus pumilus lipases from the Antarctic. J Microbiol Biotechnol 23:661–667
Bell PJ, Sunna A, Gibbs MD, Curach NC, Nevalainen H, Bergquist PL (2002) Prospecting for novel lipase genes using PCR. Microbiology 148(8):2283–2291
D’Amico S, Marx JC, Gerday C, Feller G (2003) Activity-stability relationships in extremophilic enzymes. J Biol Chem 278:7891–7896
Detry J, Rosenbaum T, Lutz S, Hahn D, Jaeger KE, Muller M, Eggert T (2006) Biocatalytic production of enantiopure cyclohexane-trans-1,2-diol using extracellular lipases from Bacillus subtilis. Appl Microbiol Biotechnol 72:1107–1116
Feller G, Gerday C (2003) Psychrophilic enzymes: hot topics in cold adaptation. Nat Rev Microbiol 1:200–208
Fields PA, Somero GN (1998) Hot spots in cold adaptation: localized increases in conformational flexibility in lactate dehydrogenase A4 orthologs of Antarctic notothenioid fishes. Proc Natl Acad Sci USA 95:11476–11481
Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol 64:763–781
Jaeger KE, Eggert T (2002) Lipases for biotechnology. Curr Opin Biotechnol 13:390–397
Jeong JY, Yim HS, Ryu JY, Lee HS, Lee JH, Seen DS, Kang SG (2012) One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl Environ Microbiol 78:5440–5443
Joseph B, Ramteke PW, Thomas G, Shrivastava N (2007) Standard review cold-active microbial lipases: a versatile tool for industrial applications. Biotechnol Mol Biol Rev 2:10
Joseph B, Ramteke PW, Thomas G (2008) Cold active microbial lipases: some hot issues and recent developments. Biotechnol Adv 26:457–470
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486
Rasool S, Johri S, Riyaz-ul-Hassan S, Maqbool QU, Verma V, Koul S, Taneja SC, Qazi GN (2005) Molecular cloning of enantioselective ester hydrolase from Bacillus pumilus DBRL-191. FEMS Microbiol Lett 249:113–120
Schmid RD, Verger R (1998) Lipases: interfacial enzymes with attractive applications. Angew Chem Int Ed Engl 37:26
Straathof AJJ (2001) Development of a computer program for analysis of enzyme kinetics by progress curve fitting. J Mol Catal B Enzy 11:991–998
van Pouderoyen G, Eggert T, Jaeger KE, Dijkstra BW (2001) The crystal structure of Bacillus subtilis lipase: a minimal alpha/beta hydrolase fold enzyme. J Mol Biol 309:215–226
Willeman WF, Hanefeld U, Straathof AJ, Heijnen JJ (2000) Estimation of kinetic parameters by progress curve analysis for the synthesis of (R)-mandelonitrile by Prunus amygdalus hydroxynitrile lyase. Enzy Microb Technol 27:423–433
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40
Acknowledgments
Experiment of Circular Dichroism spectroscopy was carried out by Mr. Hackwon Do. This research was supported by a Grant from the Ministry of Oceans and Fisheries (PM12030).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wi, A.R., Jeon, SJ., Kim, S. et al. Characterization and a point mutational approach of a psychrophilic lipase from an arctic bacterium, Bacillus pumilus . Biotechnol Lett 36, 1295–1302 (2014). https://doi.org/10.1007/s10529-014-1475-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-014-1475-8