
Abstract
The green alga, Chlamydomonas reinhardtii, is a model organism used in the study of photosynthesis and biotechnological research. Despite its importance, a complete set of genetic tools has yet to be developed. Here, we report the development of a new method for constructing a multi-gene pathway in Saccharomyces cerevisiae and integrating the assembled pathway into the nuclear genome of C. reinhardtii. To demonstrate the use of this method, we assembled and functionally expressed up to three reporter proteins (Ble, AphVIII, and GFP) simultaneously in the nucleus of C. reinhardtii. This new molecular tool should aid efforts to engineer microalgae for biofuel and biopharmaceutical production.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The eukaryotic green alga, Chlamydomonas reinhardtii, is a model organism used in the study of photosynthesis, flagellar function, and lipid metabolism (Harris 1989). Compared to other unicellular microalgae, C. reinhardtii is a particularly attractive species because of its relatively fast growth rate, simple nutrient requirements, well characterized genetics, and genetic tractability. All three genomes (nuclear, chloroplast, and mitochondrial) of C. reinhardtii have been sequenced and transformation methods that target each of these genomes have been developed (Meslet-Cladiere and Vallon 2011). Despite these advances, new genetic tools are still needed to harness the full potential of C. reinhardtii, especially in light of the surging interest in using microalgae as a source of biofuels, therapeutic proteins, fish and animal feed, and nutraceuticals (Pulz and Gross 2004).
To date, most studies have focused only on introducing single transgenes into the nuclear genome of C. reinhardtii to enhance metabolite production (Cordero et al. 2011; Leon et al. 2007; Radakovits et al. 2010). The introduction of more sophisticated biochemical pathways into the nuclear genome of C. reinhardtii has likely been hampered by the inherent limitations of traditional molecular cloning. The most complex pathway to be integrated and expressed in the nuclear genome of C. reinhardtii is the poly-3-hydroxybutyrate (PHB) biosynthetic pathway (Chaogang et al. 2010). To achieve PHB production, these authors had to co-transform C. reinhardtii with two different expression vectors containing the genes phbB and phbC. This example highlights the current difficulties of introducing multi-gene pathways into the nuclear genome of C. reinhardtii and indicates the need for a new, more robust method.
We have reported a new method for assembling and integrating biochemical pathways into the chloroplast genome of C. reinhardtii (Noor-Mohammadi et al. 2012). In this present study, we present a system based, in part, on the DNA Assembler method (Shao and Zhao 2009) for constructing and integrating multi-gene pathways into the nuclear genome of C. reinhardtii. As a proof of concept, we used this approach to assemble, integrate, and functionally express multiple reporter genes in the nucleus of C. reinhardtii.
Materials and methods
Strains and culture conditions
Chlamydomonas reinhardtii strain 137c was obtained from Life Technologies (Carlsbad, CA). The plasmid pRS426 was a gift from Dr Huimin Zhao at the University of Illinois at Urbana-Champaign. Plasmids pSP124S, pKS-aphVIII-lox, and pKScGFP were obtained from the Chlamydomonas Center (Duke University, Durham, NC). All algal cultures were grown in Tris/acetate/phosphate (TAP) medium at 23 °C under constant fluorescent white light [80 μmol m−2 s−1 as measured by a Field Scout Quantum Meter (Plainfield, IL)] on a rotary shaker at 100 rpm. Microalgae transformants were selected on 20 μg zeocin/ml or 20 μg zeocin/ml + 10 μg paromomycin/ml, when appropriate. Saccharomyces cerevisiae YSG50 was grown at 30 °C in YPAD medium containing 1 % (w/v) yeast extract, 2 % (w/v) peptone, 2 % (w/v) glucose and 0.01 % adenine hemisulphate. Escherichia coli strain DH5α was grown at 37 °C in LB medium.
Plasmid construction
Plasmids pRS426-ble, pRS426-ble-aphVIII, and pRS426-ble-aphVIII-gfp were constructed using the DNA Assembler method (Supplementary Fig. 1) (Shao and Zhao 2009). The 5′-untranslated region (UTR; rbcS2, 208 bp; β 2 -tubulin, 331 bp; psaD, 823 bp) and 3′-UTR (rbcS2, 227 bp; COP, 162 bp; psaD, 392 bp) used to construct the gene expression cassettes were PCR-amplified from the plasmids pSP124S, pKS-aphVIII-lox, and the nuclear genome of C. reinhardtii. The structural genes ble (527 bp), aphVIII (804 bp), and gfp (717 bp) were amplified from pSP124S, pKS-aphVIII-lox, and pKScGFP, respectively. Construction primers for the plasmids are listed in Supplementary Table 1. Constructed plasmids were verified to be correct restriction digestion analysis.
Nuclear transformations
For nuclear transformations, 230 μl frozen C. reinhardtii cells (GeneArt Chlamydomonas Engineering Kit, Life Technologies, Carlsbad, CA, USA) were thawed in a 37 °C water bath then mixed with 4 ml of room temperature Gibco TAP medium in a 6-well plate and grown under light for 3–6 days. Once the culture reached an OD750 >0.6, cells were diluted in fresh medium to give an OD750 of 0.06. After 20–24 h (OD750 = 0.3–0.5), 15 ml of algae culture was harvested by centrifugation at 2500×g for 10 min and then resuspended in 250 μl TAP/40 mM sucrose. 2 μg of HindIII-linearized plasmid was added to the cells and the mixture was transferred to a 4 mm electroporation cuvette and incubated at room temperature for 5 min. The following electroporation parameters were used for the transformation: 600 V, capacitance 50 μF, and resistance set to infinity. Transformed cells were mixed with 5 ml TAP/40 mM sucrose and incubated under continuous light for recovery. After 24 h, cells were harvested and resuspended in fresh TAP/40 mM sucrose and then plated on TAP/agar plates containing 20 μg zeocin/ml or 20 μg zeocin/ml + 10 μg paromomycin/ml. The plates were placed under constant light for 5–7 days until transformed algae colonies appeared. PCR analysis was performed to verify gene integration for each transgenic strain.
Functional analysis of the transgenic strains
Transgenic strains were grown in 50 ml liquid TAP medium under light with antibiotics to analyze growth. Transformants were grown in TAP media containing either 20 μg zeocin/ml (ble strains) or 20 μg zeocin/ml + 10 μg paromomycin/ml (ble-aphVIII and ble-aphVIII-gfp strains). Cell growth was monitored by measuring the OD750 using a Thermo Scientific NanoDrop 2000c and correlated to dry cell weight. GFP fluorescence of the ble-aphVIII-gfp transformants was measured using a Safire Tecan microplate reader. To analyze the GFP transformants, 3 ml cells grown to saturation were centrifuged at 17,000×g for 2 min, re-suspended in 300 μl fresh TAP medium, then 150 μl suspension was used to measure relative fluorescence using the plate reader at 488 nm excitation and 530 nm emission.
Western blot analysis
Algal cell cultures were grown to late growth phase in 40 ml of TAP medium. Cells were harvested by centrifugation and resuspended in 0.5 ml 20 mM Tris/HCl pH 8.0. The resuspended cells were lysed by sonication using a Misonix sonicator (Farmingdale, NY, USA) with the amplitude set at 30 % and with a pulse sequence of 10 s on and 10 s off, for 5 min. The insoluble cellular material was removed by centrifugation at 14,000×g for 60 min. Samples were first boiled for 7 min and separated on a 12 % SDS-PAGE and then transferred onto a methanol-treated PVDF membrane as described by Pourmir et al. (2013). The PVDF membrane was blocked with 5 % (v/v) milk in TBST buffer (20 mM Tris, 150 mM NaCl, 0.02 % Tween 20, pH 7.5) for 1 h at room temperature. The blocked membrane was incubated at 4 °C overnight in TBST solution containing 1:1000 dilution of a rabbit anti-FLAG primary antibody (Cell Signaling Technology, Danvers, MA, USA), washed three times with TBST for 5 min, incubated with 1:2000 dilution of a horseradish peroxidase conjugated anti-rabbit IgG secondary antibody (Cell Signaling Technology, Danvers, MA, USA) for 1 h, then the membrane was washed with TBST for 30 min. A working solution of Pierce ECL Substrate (Thermo Scientific, Rockford, IL, USA) was prepared according to the manufacturer’s instructions and added to the membrane for 1 min. The membrane was removed from the substrate and analyzed by an Alpha Innotech FluorChem HD2 imager and the software AlphaEaseFC.
Results and discussion
Overview of the method
An overview of the method is shown in Fig. 1. Expression cassettes consisting of a 5′-UTR, structural gene, and 3′-UTR were first assembled via overlap extension PCR (OE-PCR) (Horton et al. 2013). The expression cassettes were transformed into the yeast strain S. cerevisiae YSG50, where in vivo homologous recombination is used to assemble the biochemical pathway into the vector pRS426. The assembled plasmid was isolated from yeast then transformed into the nuclear genome of C. reinhardtii.

Overview of the method. Assembly of the biochemical pathway occurs via homologous recombination in S. cerevisiae. The assembled plasmid is isolated from yeast, then transformed into the microalga C. reinhardtii where it integrates into the nuclear genome
Construction of the expression cassettes
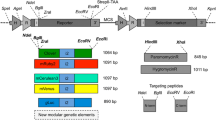
Expression of both homologous and heterologous genes in the nucleus of C. reinhardtii is heavily influenced by promoter and regulatory elements. The most commonly used promoters for nuclear expression are those from the rbcS2, psaD, HSP70A, and β 2 -tubulin genes (Griesbeck et al. 2006; Specht et al. 2010). High levels of transcription and expression have also been achieved using chimeric constitutive promoters and by placing endogenous intronic sequences in transgenes (Lumbreras et al. 1998). Inducible promoters, such as NIT1, CA1, and CYC6, that allow for more controllable expression have been developed (Ferrante et al. 2008). For this work, we selected the rbcS2, psaD, and β 2 -tublin promoters. A unique 5′-UTR and 3′-UTR was chosen to flank each gene to minimize the potential for unintentional homologous recombination during assembly in yeast. Expression cassettes consisting of a 5′-UTR, gene (ble, aphVIII, or gfp), and 3′-UTR were PCR-amplified, then assembled using OE-PCR. Assembled expression cassettes along with linearized pRS426 were co-transformed into yeast. The resulting fully assembled vectors contained the one-gene, two-gene, and three-gene pathways (Fig. 2). Correct assembly of these three vectors was confirmed by restriction digestion analysis (Supplementary Fig. 2).

Expression cassettes used for heterologous gene expression from the nuclear genome of C. reinhardtii. Expression cassettes were each assembled into the vector pRS426. Genes are flanked by 5′- and 3′-untranslated regions. Constructs used in this study include: a pRS426-ble, b pRS426-ble-aphVIII, and c pRS426-ble-aphVIII-gfp
Integration into the nuclear genome of C. reinhardtii
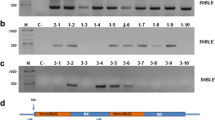
The assembled plasmids pRS426-ble, pRS426-ble-aphVIII, and pRS426-ble-aphVIII-gfp were each linearized by restriction enzyme digestion and transformed into C. reinhardtii by electroporation. Transformants were selected on TAP plates supplemented with the antibiotic zeocin and screened by PCR to verify integration of each of the genes (Supplementary Fig. 3). Transformation efficiencies for the three constructs are shown in Table 1. The % gene-positive clones identified seemed to decrease with increasing pathway size.
Functional analysis of the engineered strains
The functional expression of the integrated ble, ble-aphVIII, and ble-aphVIII-gfp pathways were analyzed by growing the transformant strains in batch cultures of TAP medium supplemented with either zeocin or zeocin plus paromomycin. As shown in Fig. 3a, the ble transformant was able to grow in the presence of the antibiotic zeocin, whereas wild-type C. reinhardtii did not grow. Similarly, the ble-aphVIII and ble-aphVIII-gfp transformant strains grew in the presence of zeocin plus paromomycin, while no growth was observed from wild-type C. reinhardtii (Fig. 3b). These results demonstrate the functional expression of the ble and aphVIII genes in the engineered strains of C. reinhardtii.

Growth of the engineered strains in TAP medium supplemented with the antibiotics a zeocin and b zeocin + paromomycin. Wild-type C. reinhardtii served as a negative control
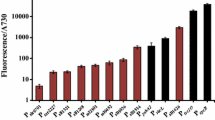
To analyze GFP expression, six clones of ble-aphVIII-gfp were randomly chosen and the fluorescence of each of these clones was measured using a microplate reader. Six clones were chosen for analysis because heterologous genes have been shown to be inserted randomly into the nuclear genome of C. reinhardtii, resulting in a wide range of heterologous gene expression levels (Slaninová et al. 2008). Chlamydomonas naturally produces a variety of highly fluorescent pigments, such as chlorophylls and flavonoids, which results in a high level of auto-fluorescence (Rasala et al. 2013), thus the clones were analyzed relative to wild-type C. reinhardtii. As shown in Fig. 4a, fluorescence above auto-fluorescence was detected in four of the six clones tested (C1, C2, C4, and C5). The variability of fluorescence across the four clones ranged from 1.1 to 1.4 times more fluorescence than wild-type C. reinhardtii.

a Quantification of the relative GFP fluorescence of six selected ble-aphVIII-gfp clones (C1-C6). The ble-aphVIII-gfp clones expressing GFP are shown relative to wild-type (WT) C. reinhardtii. b Western blot analysis of the six ble-aphVIII-gfp clones. WT C. reinhardtii and a 16S/GFP C. reinhardtii chloroplast transformant (unpublished work) were included as negative and positive controls, respectively
In addition to growth and fluorescence analysis, the ble-aphVIII-gfp clones were also subjected to western blot analysis to detect GFP protein accumulation in the nucleus of C. reinhardtii. As shown in Fig. 4b, three of the clones (C1, C2, and C5) had clearly discernible bands indicating GFP accumulation. Interestingly, despite displaying a 10 % increase in relative fluorescence compared to wild-type (Fig. 4a), the clone C4 did not have a detectable GFP band on the western blot; however, the other three clones seem to show a strong correlation between relative fluorescence and GFP band intensity.
Conclusions
This study presents the design and application of a new system for expressing biochemical pathways in the nucleus of C. reinhardtii. Using this method, it should be possible to introduce larger and more complex biosynthetic pathways into the nuclear genome of C. reinhardtii and to optimize the expression of each gene in the pathway to maximize the production of a target compound. This new molecular tool should be useful for engineering microalgae for biofuel and biotechnological applications.
References
Chaogang W, Zhangli H, Anping L, Baohui J (2010) Biosynthesis of poly-3-hydroxybutyrate (PHB) in the transgenic green alga Chlamydomonas reinhardtii. J Phycol 46:396–402
Cordero BF, Couso I, Leon R, Rodriguez H, Vargas MA (2011) Enhancement of carotenoids biosynthesis in Chlamydomonas reinhardtii by nuclear transformation using a phytoene synthase gene isolated from Chlorella zofingiensis. Appl Microbiol Biotechnol 91(2):341–351
Ferrante P, Catalanotti C, Bonente G, Giuliano G (2008) An optimized, chemically regulated gene expression system for Chlamydomonas. PLoS One 3:e3200. doi:10.1371/journal.pone.0003200
Griesbeck C, Kobl I, Heitzer M (2006) Chlamydomonas reinhardtii: a protein expression system for pharmaceutical and biotechnological proteins. Mol Biotechnol 34(2):213–223
Harris EH (1989) The Chlamydomonas sourcebook: a comprehensive guide to biology and laboratory use. Academic Press, San Diego
Horton RM, Cai Z, Ho SM, Pease LR (2013) Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 54:129–133
Leon R, Couso I, Fernandez E (2007) Metabolic engineering of ketocarotenoids biosynthesis in the unicelullar microalga Chlamydomonas reinhardtii. J Biotechnol 130:143–152. doi:10.1016/j.jbiotec.2007.03.005
Lumbreras V, Stevens DR, Purton S (1998) Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J 14:441–447
Meslet-Cladiere L, Vallon O (2011) Novel shuttle markers for nuclear transformation of the green alga Chlamydomonas reinhardtii. Eukaryot Cell 10:1670–1678
Noor-Mohammadi S, Pourmir A, Johannes TW (2012) Method to assemble and integrate biochemical pathways into the chloroplast genome of Chlamydomonas reinhardtii. Biotechnol Bioeng 109:2896–2903
Pourmir A, Noor-Mohammadi S, Johannes TW (2013) Production of xylitol by recombinant microalgae. J Biotechnol 165:178–183
Pulz O, Gross W (2004) Valuable products from biotechnology of microalgae. Appl Microbiol Biotechnol 65:635–648
Radakovits R, Jinkerson RE, Darzins A, Posewitz MC (2010) Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell 9:486–501
Rasala BA, Barrera DJ, Ng J, Plucinak TM, Rosenberg JN, Weeks DP, Oyler GA, Peterson TC, Haerizadeh F, Mayfield SP (2013) Expanding the spectral palette of fluorescent proteins for the green microalga Chlamydomonas reinhardtii. Plant J 74(4):545–556
Shao Z, Zhao H (2009) DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res 37:e16. doi:10.1093/nar/gkn991
Slaninová M, Hroššová D, Vlček D, Mages W (2008) Is it possible to improve homologous recombination in Chlamydomonas reinhardtii? Biologia 63:941–946
Specht E, Miyake-Stoner S, Mayfield S (2010) Micro-algae come of age as a platform for recombinant protein production. Biotechnol Lett 32:1373–1383
Acknowledgments
This work was supported by a Grant from the U.S. Department of Energy (DE-SC0005315). S.N. and A.P. were supported by Conoco-Phillips fellowships.
Author information
Authors and Affiliations
Corresponding author
Additional information
Samaneh Noor-Mohammadi and Azadeh Pourmir contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Noor-Mohammadi, S., Pourmir, A. & Johannes, T.W. Method for assembling and expressing multiple genes in the nucleus of microalgae. Biotechnol Lett 36, 561–566 (2014). https://doi.org/10.1007/s10529-013-1378-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-013-1378-0