
Abstract
The measurements of plasma natriuretic peptides (NT-proBNP, proBNP and BNP) are used to diagnose heart failure but these are expensive to produce. We describe a rapid, cheap and facile production of proteins for immunoassays of heart failure. DNA encoding N-terminally His-tagged NT-proBNP and proBNP were cloned into the pJexpress404 vector. ProBNP and NT-proBNP peptides were expressed in Escherichia coli, purified and refolded in vitro. The analytical performance of these peptides were comparable with commercial analytes (NT-proBNP EC50 for the recombinant is 2.6 ng/ml and for the commercial material is 5.3 ng/ml) and the EC50 for recombinant and commercial proBNP, are 3.6 and 5.7 ng/ml respectively). Total yield of purified refolded NT-proBNP peptide was 1.75 mg/l and proBNP was 0.088 mg/l. This approach may also be useful in expressing other protein analytes for immunoassay applications.
Purpose of work
To develop a cost effective protein expression method in E. coli to obtain high yields of NT-proBNP (1.75 mg/l) and proBNP (0.088 mg/l) peptides for immunoassay use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a global health problem associated with poor clinical outcomes and substantial economic burden to our healthcare system. (Dunlay et al. 2011). Approx. 23 million people worldwide are living with HF and this figure is likely to increase in the near future due to an ageing and growing population (Cheng and Vasan 2011). The population estimates of HF prevalence ranges between 2 and 10 %, with a higher prevalence in the elderly (Manzano et al. 2011). The diagnosis of HF is challenging and is currently based on a combination of patient medical history, physical examinations coupled with routine clinical procedures (Krum et al. 2011).
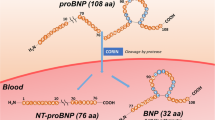
Measurement of plasma N-terminal pro-brain natriuretic peptide (NT-proBNP) for diagnosis of HF is now clinically accepted by the major HF guidelines (Krum et al. 2011; McMurray et al. 2012; Lindenfeld et al. 2010). During the onset of HF, the 134 amino acid (AA) precursor protein (preproBNP) is predominantly expressed in cardiomyocytes and is secreted into circulation (Dong et al. 2010). The signal peptide corresponding to the first 26 AA of preproBNP is cleaved, resulting in the production of the 108 AA pro B-type natriuretic protein (proBNP). ProBNP is cleaved by the protease corin that is present in circulation into a 76 AA NT-proBNP fragment and 32 AA BNP (Nishikimi et al. 2011; Semenov and Seferian 2011). Low levels of proBNP and/or NT-proBNP are also present in the circulation of the healthy population (Fradley et al. 2011; Tonne et al. 2011). HF patients tend to have a less efficient mechanism of converting proBNP into NT-proBNP and BNP (Semenov et al. 2010; Emdin et al. 2011) and therefore proBNP may be clinically relevant when diagnosing HF.
NT-proBNP is present in other biological fluids. The use of urinary NT-proBNP measurements is also clinically useful in diagnosing HF (Palmer et al. 2009). Furthermore, we have demonstrated diagnostic potential of this NT-proBNP in an alternative biological fluids (Zhang et al. 2013; Foo et al. 2013; Schulz et al. 2013). Nevertheless, it is important that the development of future NT-proBNP and proBNP immunoassays validate these findings.
The cost of purchasing an analyte as a calibrant is a major hindrance when developing new immunoassays to detect proteins present in cells, tissues and body fluids. The expression and purification of recombinant NT-proBNP has been described (Ala-Kopsala et al. 2004, 2005). However, this method used the production of antisera against NT-proBNP that required the removal of N-terminally GST-tagged NT-proBNP through thrombin cleavage. In contrast, here we describe a rapid, cheap and facile protein expression method for N-terminally His-tagged NT-proBNP and proBNP in an Escherichia coli system with high yields (1.75 mg NT-proBNP/l and 0.088 mg proBNP/l) for downstream immunoassay development.
Materials and methods
Sequence and cloning of N-terminally His-tagged preproBNP
DNA sequence encoding N-terminally His-tagged preproBNP optimized for expression in E. coli was synthesized and cloned into the pJexpress404 vector (DNA2.0) (Fig. 1a). Using the PCR deletion method of Imai et al. (1991), we deleted sequence encoding the signal peptide, pro-piece and/or BNP to create plasmids encoding cytoplasmically targeted, N-terminally His-tagged proBNP (Fig. 1b) and NT-proBNP (Fig. 1c). Forward (5′-CAT CAC CAC CAC CAT CAT CAC-3′) and reverse primers (5′-CAT ATG TAT ATC TCC TTC-3′) were used to delete sequence encoding the signal peptide. Forward (5′-TAA CTC GAG CCC CAA GGG-3′) and reverse primers (5′-ACG CGG TGC ACG CAG GG-3′) were used to delete the sequence encoding BNP (Fig. 1a).

Sequence of E. coli expressed NT-proBNP and proBNP. a Protein and E. coli optimized DNA sequence of His-tagged proBNP with signal sequence. Oligonucleotide primer sequences are underlined. b Protein sequence of N-terminally His-tagged proBNP after sequence encoding signal peptide was deleted (blue) c Protein sequence of N-terminally His-tagged NT-proBNP after deletion of sequence encoding signal peptide (blue) and BNP (red)
Expression and purification of recombinant NT-proBNP and proBNP using an E. coli system
Escherichia coli with the desired plasmids encoding proBNP or NT-proBNP was grown in 400 ml LB with 100 mg ampicillin/l at 37 °C with shaking. When the cultures reached an OD600 of 0.7, IPTG was added at 1 mM to induce protein expression and the cultures were incubated for an additional 4 h at 37 °C with shaking. The cells were harvested by centrifugation. Harvested cells containing proBNP or NT-proBNP inclusion bodies were resuspended in 6 M guanidine hydrochloride with 50 mM potassium phosphate buffer, pH 7.4, and incubated at 37 °C for 16 h. proBNP or NT-proBNP were purified using TALON resin affinity chromatography (Clontech) according to the manufacturer’s instructions, with elution in 6 M guanidine hydrochloride with 50 mM potassium phosphate buffer pH 5.
Desalting and measurement of in-house expressed analytes
ProBNP and NT-proBNP were refolded by dilution to 1 M guanidine hydrochloride with 50 mM potassium phosphate buffer (pH 7.4) and incubated at 4 °C for 4 h. The recombinant proBNP or NT-proBNP were desalted into 10 mM potassium phosphate buffer pH 7.4 with 150 mM NaCl using two PD-10 columns (14.5 × 50 mm) at 4 °C. The protein concentration of recombinant NT-proBNP was measured using a protein quantification kit and proBNP measured using NanoDrop. Recombinant proBNP and NT-proBNP were aliquoted and stored at −80 °C until further analysis.
In-house expressed NT-proBNP and proBNP characterization using mass spectrometry
Recombinant NT-proBNP and proBNP were separated by bis–Tris SDS-PAGE and stained with Coomassie Blue according to the manufacturer’s instructions. Purified proteins were digested with trypsin in 50 mM Tris/HCl pH 7.5 with 10 mM DTT at 37 °C for 16 h. Tryptic peptides were desalted using C18 ZipTips (Millipore) and analysed by LC–ESI–MS/MS using a Prominence nano LC system (Shimadzu) and TripleTof 5600 mass spectrometer with a Nanospray III interface (AB Sciex) (Bailey et al. 2012a). Approx. 2 μg peptides were desalted on an Agilent C18 trap (300 Å pore size, 5 μm particle size, 0.3 mm i.d. × 5 mm) at 30 μl/min for 3 min, and then separated on a reversed-phase C18 HPLC column (300 Å pore size, 5 μm particle size, 150 μm i.d. × 150 mm) at 1 μl/min. Peptides were separated with a gradient of 10–60 % buffer B over 45 min, with buffer A (1 % acetonitrile/0.1 % formic acid) and buffer B (80 % acetonitrile/0.1 % formic acid). Gas and voltage setting were adjusted as required. A MS TOF scan from m/z of 350–1,800 was performed for 0.5 s followed by information dependent acquisition of MS/MS with automated CE selection of the top 20 peptides from m/z of 40–1,800 for 0.05 s per spectrum.
Data analysis
Peptides were identified essentially as described by Bailey et al. (2012a) using ProteinPilot (AB Sciex), searching the LudwigNR database (downloaded from http://apcf.edu.au as at 27 January 2012; 16,818,973 sequences; 5,891,363,821 residues) with standard settings [sample type, identification; cysteine alkylation, acrylamide; instrument, TripleTof 5600; species, no restriction; ID focus, biological modifications; enzyme, trypsin; Search effort, thorough ID]. False discovery rate analysis using ProteinPilot was performed on all searches. Peptides identified with greater than 99 % confidence and with a local false discovery rate of less than 1 % were included for further analysis, and MS/MS fragmentation spectra were manually inspected.
Suitability of recombinant NT-proBNP and proBNP as calibrants for AlphaLISA immunoassays
To determine the suitability of the recombinant proBNP and NT-proBNP peptides as analytes in immunoassays, 12-point standard curves were generated by serial dilution of known concentrations of recombinant proBNP and recombinant NT-proBNP. The standard curves were generated by plotting the “raw” AlphaLISA counts against the proBNP or NT-proBNP concentrations using a 4-parameter logistic equation (sigmoidal dose–response curve with variable slope) and a 1/Y2 data weighting using GraphPad Prism 5 software version 5.03 (GraphPad Software Inc., USA) (Punyadeera et al. 2011; Mohammed et al. 2012; Topkas et al. 2012; Bailey et al. 2012b). The recombinantly expressed analytes (proBNP and NT-proBNP) were directly compared with a commercial proBNP and NT-proBNP analyte (Product-No: 1607106, Perkin Elmer, MA, USA) in two separate AlphaLISA immunoassays.
Stability study of our in-house expressed recombinant and commercial proBNP and NT-proBNP in HiBlock buffer was performed by storing several aliquots of the proteins at 4 °C. An aliquot of the recombinant and commercial proBNP and NT-proBNP were then stored at −80 °C at specific time intervals (1, 3 and 5 days) before further analysis. AlphaLISA assays were performed and percentage recoveries were calculated based on concentration of the recombinant proteins at day 0.
Results
SDS-PAGE validation of in-house expressed and purified recombinant proBNP and NT-proBNP peptides
We used an E. coli heterologous protein expression system to express, purify and refold human proBNP and NT-proBNP. A critical step in the development of immunoassays is the quality of the analyte used as a calibrant, thus SDS-PAGE followed by Coomassie Blue staining was also performed to characterise the recombinant proBNP and NT-proBNP analytes produced in-house.
The molecular sizes of recombinantly expressed proBNP and NT-proBNP were approx. 15 and 9 kDa respectively, in agreement with the expected molecular sizes. Single high intensity bands were observed in the lanes loaded with the respective recombinant analytes, which were >95 % pure (Fig. 2a).

SDS-PAGE and MS characterisation of purified NT-proBNP and proBNP. a Approx. 1 μg of purified recombinant NT-proBNP and proBNP were loaded in lane 1 and 2, respectively. The proteins were separated by SDS-PAGE and stained with Coomassie blue. MS peptide coverage of purified b NT-proBNP and c proBNP. Sequences shown in bold are tryptic peptides identified with >99 % confidence (see Tables 1, 2)
Mass spectrometry analysis of in-house expressed recombinant proBNP and NT-proBNP
Peptides from proBNP and NT-proBNP were detected by LC–MS/MS (Fig. 2b–c; Tables 1, 2). NT-proBNP peptides included the N-terminus and C-terminus of the full-length of NT-proBNP, indicating that our recombinant protein is not subject to any N- or C-terminal truncations, and that the protein has the expected sequence throughout (Table 1; Fig. 2b). Peptides from proBNP were also detected from throughout the full length of the molecule (Table 2; Fig. 2c). A peptide from the centre of proBNP and NT-proBNP was identified which is glycosylated in vivo and was not reported in previous publications describing MS analysis of immunoprecipitation (IP) purified NT-proBNP from patient’s plasma (Hammerer-Lercher et al. 2008).
Validation of in-house expressed proBNP and NT-proBNP in AlphaLISA assays
As guanidine hydrochloride is not compatible with the AlphaLISA assay, refolded NT-proBNP or proBNP were desalted into 10 mM potassium phosphate buffer pH 7.4 with 150 mM NaCl. The in-house expressed recombinant NT-proBNP yielded 3.5 ml of analyte at approx. 1.75 mg/l bacterial culture. In addition, expression of recombinant proBNP gave approx. 0.088 mg/l bacterial culture. The analytical sensitivity and specificity of our purified N-terminally His-tagged NT-proBNP were compared with a commercial analyte as a calibrant in a NT-proBNP immunoassay.
Comparable 12-point sigmoidal-dose response curves with similar detection ranges between 10 and 100,000 pg/ml were obtained (EC50 = 3.6 ng/ml for proBNP and EC50 = 2.6 ng/ml for NT-proBNP) with both in-house expressed recombinant proBNP and NT-proBNP analytes (Fig. 3a, b). In addition, we directly compared in-house expressed proBNP and NT-proBNP to commercial analytes and found similar EC50 values (EC50 = 5.7 ng/ml and EC50 = 3.6 ng/ml for commercial and in-house expressed recombinant proBNP analytes respectively; EC50 = 5.3 ng/ml and EC50 = 2.6 ng/ml for commercial and in-house expressed recombinant NT-proBNP analyte respectively), supporting the suitability of our recombinant proBNP and NT-proBNP as a standard/calibrant for use in immunoassays. The assay sensitivity for our in- house expressed proBNP lower limit of detection (LOD) = 20 pg/ml and NT-proBNP LOD = 16 pg/ml.

(a, b) Binding saturation curve of recombinant and commercial proBNP and NT-proBNP to monoclonal antibodies. (b, c) Stability (0, 1, 3 and 5 days) of recombinant and commercial proBNP and NT-proBNP in HiBlock buffer at 4 °C measured in percentage recoveries of concentration referenced to day 0
We have demonstrated that the recombinant and commercial proBNP and NT-proBNP are stable at 4 °C (Fig. 3c, d). This confirmed that the recombinant proBNP and NT-proBNP proteins are stable and functional and that the presence of an N-terminal His-tag did not alter the immunoreactivity of proBNP and NT-proBNP in the AlphaLISA assay.
Discussion
Our work demonstrates a cost-effective method to obtain high yields of recombinant proBNP and NT-proBNP proteins (0.088 and 1.75 mg/l respectively) in a research laboratory setting. We have demonstrated that our in-house expressed analytes are compatible with downstream immunoassays (Fig. 3). Although Ala-Kopsala et al. (2004, 2005) previously described the expression and purification of recombinant NT-proBNP, their method used the production of antisera against NT-proBNP and required the use of an N-terminal GST-tag on NT-proBNP for purification and its subsequent removal through thrombin cleavage. In comparison, we have described expression of N-terminally His-tagged NT-proBNP that does not require affinity tag cleavage prior to use in an immunoassay making it a versatile method. Protein characterisation by SDS-PAGE and mass spectrometry of recombinant proBNP and NT-proBNP further confirmed their purity (Tables 1, 2; Fig. 2). It is important to validate the sequence of a recombinant proBNP and NT-proBNP analytes, as the amino and carboxyl terminal of circulating NT-proBNP is more susceptible to proteolytic degradations (Foo et al. 2013) and could lead to an underestimation of analyte levels when used in immunoassays that target both termini (N- and C) of the analyte (Ala-Kopsala et al. 2004).
The AlphaLISA results demonstrated that our recombinant peptides have equivalent immunoreactivities to commercial material (Fig. 3), indicating that the presence of an N-terminal His-tag on NT-proBNP did not interfere with the performance of immunoassay. The estimated material cost starting from designing the preproBNP plasmid until the verification of recombinant NT-proBNP and proBNP was approx. 1,000 AUD, which yielded NT-proBNP (1 mg/l 95 % pure analyte/l) and proBNP (0.088 mg 95 % pure analyte/l). Additional expression would not require gene synthesis and cloning and so would cost approx. 100 AUD per 1 mg NT-proBNP/l and 0.088 mg proBNP/l. In contrast, 1 mg of commercial NT-proBNP costs approx. $ (Australian) 4,500–15,000 (http://www.sunny-lab.com, http://www.mybiosource.com). Also, 1 mg commercial recombinant proBNP is $ (Australian) ~27,000–59,000 (http://www.mybiosource.com). By inducing NT-proBNP and proBNP expressing E. coli cells from glycerol stocks, we are able to rapidly obtain recombinant NT-proBNP and proBNP for future applications. Thus, this method will provide substantial cost savings to laboratory researchers.
Conclusion
We have optimised an E. coli expression system for the production of recombinant human proBNP and NT-proBNP. Recombinant proBNP and NT-proBNP with a purity of 95 % could be used as calibrants in an AlphaLISA immunoassay with similar immunoreactivity to commercial analyte. The cloning and purification of recombinantly expressed analyte described in our study is cost effective and we anticipate that similar approaches will also be applicable for expression of other recombinant analytes for immunoassay development.
The cost of purchasing an analyte as a calibrant is a major hindrance when developing new immunoassays to detect proteins present in cells, tissues and body fluids. The ability to rapidly produce cheap recombinant proteins to be used as calibrants in assays will facilitate assay development. Lower analyte costs allow funding to be allocated to the development and optimisation of the assay.
References
Ala-Kopsala M, Magga J, Peuhkurinen K, Leipala J et al (2004) Molecular heterogeneity has a major impact on the measurement of circulating N-terminal fragments of A- and B-type natriuretic peptides. Clin Chem 50:1576–1588
Ala-Kopsala M, Ruskoaho H, Leppaluoto J, Seres L et al (2005) Single assay for amino-terminal fragments of cardiac A- and B-type natriuretic peptides. Clin Chem 51:708–718
Bailey UM, Jamaluddin MF, Schulz BL (2012a) Analysis of congenital disorder of glycosylation-Id in a yeast model system shows diverse site-specific under-glycosylation of glycoproteins. J Proteome Res 11:5376–5383
Bailey UM, Punyadeera C, Cooper-White JJ, Schulz BL (2012b) Analysis of the extreme diversity of salivary alpha-amylase isoforms generated by physiological proteolysis using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 911:21–26
Cheng S, Vasan RS (2011) Advances in the epidemiology of heart failure and left ventricular remodelling. Circulation 124:516–519
Dong N, Chen S, Yang J, He L et al (2010) Plasma soluble corin in patients with heart failure. Circul Heart Fail 3:207–211
Dunlay SM, Shah ND, Shi Q, Morlan B et al (2011) Lifetime costs of medical care after heart failure diagnosis. Circ Cardiovasc Qual Outcomes 4:68–75
Emdin M, Passino C, Clerico A (2011) Natriuretic peptide assays revisited: do we need pro-B-type natriuretic peptide? J Am Coll Cardiol 57:1396–1398
Foo JY, Wan Y, Schulz BL, Kostner K et al. (2013) Circulating fragments of N-terminal pro B-type natriuretic peptide in plasma of heart failure patients. Clin Chem
Fradley MG, Larson MG, Cheng S, McCabe E et al (2011) Reference limits for N-terminal-pro-B-type natriuretic peptide in healthy individuals (from the Framingham heart study). Am J Cardiol 108:1341–1345
Hammerer-Lercher A, Halfinger B, Sarg B, Mair J et al (2008) Analysis of circulating forms of proBNP and NT-proBNP in patients with severe heart failure. Clin Chem 54:858–865
Imai Y, Matsushima Y, Sugimura T, Terada M (1991) A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res 19:2785
Krum H, Jelinek MV, Stewart S, Sindone A et al (2011) 2011 update to National Heart Foundation of Australia and Cardiac Society of Australia and New Zealand Guidelines for the prevention, detection and management of chronic heart failure in Australia, 2006. Med J Aust 194:405–409
Lindenfeld J, Albert NM, Boehmer JP, Collins SP et al (2010) HFSA 2010 Comprehensive heart failure practice guideline. J Card Fail. 16:e1–e194
Manzano L, Babalis D, Roughton M, Shibata M et al (2011) Predictors of clinical outcomes in elderly patients with heart failure. Eur J Heart Fail 13:528–536
McMurray JJ, Adamopoulos S, Anker SD, Auricchio A et al (2012) ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 33:1787–1847
Mohammed R, Leigh Cambell J, Cooper-White J, Dimesky G et al (2012) The impact of saliva collection and processing methods on Crp, Ige, and Myoglobin immunoassays. Clin Transl Med 1:19
Nishikimi T, Kuwahara K, Nakao K (2011) Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J Cardiol 57:131–140
Palmer SC, Endre ZH, Richards AM, Yandle TG (2009) Characterization of NT-proBNP in human urine. Clin Chem 55:1126–1134
Punyadeera C, Dimeski G, Kostner K, Beyerlein P et al (2011) One-step homogeneous C-reactive protein assay for saliva. J Immunol Methods 373:19–25
Schulz BL, Cooper-White J, Punyadeera CK (2013) Saliva proteome research: current status and future outlook. Crit Rev Biotechnol 33:246–259
Semenov AG, Seferian KR (2011) Biochemistry of the human B-type natriuretic peptide precursor and molecular aspects of its processing. Clin Chim Acta 412:850–860
Semenov AG, Tamm NN, Seferian KR, Postnikov AB et al (2010) Processing of pro-B-type natriuretic peptide: furin and corin as candidate convertases. Clin Chem 56:1166–1176
Tonne JM, Campbell JM, Cataliotti A, Ohmine S et al (2011) Secretion of glycosylated pro-B-type natriuretic peptide from normal cardiomyocytes. Clin Chem 57:864–873
Topkas E, Keith P, Dimeski G, Cooper-White J et al (2012) Evaluation of saliva collection devices for the analysis of proteins. Clin Chim Acta 413:1066–1070
Zhang X, Wan Y, Cooper-White J, Goce D et al. (2013) Quantification of D-dimer levels in human saliva. Bioanalysis (in press)
Acknowledgments
The authors would like to acknowledge the financial support of the Queensland Government Smart Futures Fellowship Programme (QGSFF), the University of Queensland New Staff Research Funds (UQNSRSF 601252), University of Queensland Foundation Research Excellence Award Scheme, National Health and Medical Research Council Project Grant 631615 and National Health and Medical Research Council Career Development Fellowship APP1031542.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soleh, M.T., Foo, J.Y.Y., Bailey, UM. et al. A rapid and cost-effective method of producing recombinant proBNP and NT-proBNP variants in Escherichia coli for immunoassay of heart failure. Biotechnol Lett 36, 133–140 (2014). https://doi.org/10.1007/s10529-013-1341-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-013-1341-0