
Abstract
The vpl2 gene, encoding versatile peroxidase (VP) from Pleurotus eryngii, was synthesized with codon optimization and cloned into vector-pET-32a(+) and over-expressed in Escherichia coli BL21(DE3). An active peroxidase fused to the thioredoxin–hexahistidine tag was directly obtained by IPTG induction in the presence of hemin. Most of over-expressed protein was in the soluble form, and was purified on a nickel column with >85 % purity at a yield of 12.5 mg/l. The purified fusion protein, having an Rz value (A407/A280, a measure of hemin content of the peroxidases) of 1.2, oxidized ABTS veratryl alcohol, Mn2+, and Reactive Black 5. Activity of the enzyme increased after removing the tag. It lost only 5 % of its activity in 6.4 mM H2O2. This is the first report on direct over-expression of active VP in E. coli.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lignin-degrading peroxidases can oxidize a variety of aromatic compounds including high-redox-potential compounds such as lignin, industrial dyes, pesticides, etc. (Martínez et al. 2009). Four types of lignin-modifying enzymes from the white-rot fungus have been identified and characterized: lignin peroxidase (LiP), manganese peroxidase (MnP), versatile peroxidase (VP), and laccase (Martínez et al. 2009). Among them, laccases belong to low-redox-potential oxidoreductases, which can only oxidize high-redox-potential substrates in the presence of redox mediators, whereas the other three belong to high-redox-potential peroxidases, which can directly oxidize high-redox-potential substrates and/or Mn2+. VP presents special biotechnological interests due to its catalytic versatility (Martínez et al. 2009).
VP has been described in several species from the genera Pleurotus and Bjerkandera sharing the catalytic properties of LiP and MnP (Martínez et al. 1996; Mester and Field 1998). The crystal structure of VP from Pleurotus eryngii, shows a hybrid molecular architecture that includes different catalytic sites for oxidation of Mn2+ and high-redox-potential aromatic compounds in the same protein, which are closely related to those found in the crystal structures of MnP and LiP (Pérez-Boada et al. 2005). The exposed Trp 164 in VP is involved in long-range electron transfer for oxidation of high-redox-potential compounds like recalcitrant lignin (Pérez-Boada et al. 2005). VP follows similar catalytic cycle to other fungal peroxidases, including LiP and MnP, except that VP is unique regarding the substrates that it is able to oxidize (Pérez-Boada et al. 2005).
The first successful example to produce a fungal ligninolytic peroxidase is that of MnP which was expressed in baculovirus, but only giving a low yield of protein (Pease et al. 1991). Expression of MnP in Phanerochaete chrysosporium has been achieved (Mayfield et al. 1994); however, the main challenges involve contamination from the wild-type enzyme and extensive screening productive clones. MnP and LiP were also heterologously over-expressed in Escherichia coli, but the recombinant proteins usually accumulate as an insoluble form in the cytoplasmic inclusion bodies (Doyle and Smith 1996; Whitwam et al. 1995). It is not easy to obtain an active enzyme at high yield even after optimizing the refolding conditions, only a refolding yield less than 7 % was achieved for P. eryngii VP expressed in E. coli (Pérez-Boada et al. 2002). VP was also expressed in the ascomycete Emericella nidulans, but this approach demanded extensive screening and provided different glycosylation from the original fungus (Lu-Chau et al. 2004). Over-expression of soluble Bjerkandera adusta VP has been achieved in E. coli under auto-induction conditions in the presence of hemin, but a complicated procedure was required (Mohorčič et al. 2009). A fusion VP from P. eryngii has been produced up to 21 mg/l in Saccharomyces cerevisiae after six rounds of directed evolution (Garcia-Ruiz et al. 2012).
Production of large quantities of enzymes with an industrial interest is a key for their industrial use. In the present paper we report direct overexpression of active P. eryngii VP in E. coli under IPTG induction in the presence of hemin, characterization, and investigation into H2O2 stability.
Materials and methods
Materials
Chemicals were from Sigma, Merck or Ameresco. Oligonucleotides and vpl2 gene encoding VP from P. eryngii with codon optimization were synthesized by Shanghai Sangon Biotech Co. Ltd (China). Taq DNA polymerases and all restriction endonucleases were from Fermentas or Takara Biotechnology. The kits used for molecular cloning were from Omega Bio-tek or Takara Biotechnology. Nickel column and expression vectors were from Novagen. Antibodies and chemical reagents used for western blot were from Tiangen (China). Recombinant bovine enterokinase was from Shanghai Sangon Biotech Co. Ltd (China).
Bacterial strains, plasmids, and media
Escherichia coli DH5α was used for routine DNA transformation and plasmid isolation. E. coli BL21(DE3) was utilized for VP over expression. E. coli strains were routinely grown in Luria–Bertani broth at 37 °C with aeration or on LB supplemented with 1.5 % (w/v) agar. 50 µg ampicillin/ml or 15 μg kanamycin/ml was added when required. Vectors-pET-32a(+) and pET-28a(+) were used for subcloning.
DNA manipulations
General molecular biology techniques were carried out by standard procedures. Restriction and modification enzymes were used following the recommendations of the manufacturers. DNA fragments were purified from agarose gels using the DNA gel extraction kit. Plasmid DNA was isolated using the plasmid miniprep kit.
The vpl2 gene was amplified with forward primer (CTTCGGAATTCGCCACTTGTGACGATGGTCGAAC, EcoRI restriction site underlined) and reverse primer (AATATTGCGGCCGCTTATGATCCGGGAACTGGAGG, NotI restriction site underlined). For PCR, 50 ng plasmid DNA (vpl cloned into pBluescript II SK plus) was used as template in 50 μl reaction mixture containing PCR buffer (10 mM Tris/HCl, pH 8.8, 50 mM KCl, 0.08 % NP-40), 2 mM MgCl2, 0.5 μM each of forward and reverse primers and two units of Taq DNA polymerase. PCR cycling profiles were 1 cycle at 95 °C for 5 min, 35 cycles of 95 °C for 30 s, 56 °C for 30 s and 72 °C for 90 s, followed by a final extension step at 72 °C for 10 min. The PCR product was isolated by agarose electrophoresis and extracted from agarose gel using the DNA gel extraction kit, digested with restriction enzymes EcoRI and NotI, re-cloned into the vectors-pET-32a(+) and pET-28a(+) digested with EcoRI and NotI, respectively. All constructs were confirmed by DNA sequencing.
Protein over-expression and purification
The expression constructs pET32a-VP and pET28a-VP were transformed into competent E. coli BL21(DE3) and expressed under IPTG induction respectively. IPTG induction was carried out following the standard procedure. A single colony was used to inoculate 5 ml LB medium containing 50 μg ampicillin/ml or 15 μg kanamycin/ml and incubated at 225 rpm and 37 °C overnight. The overnight culture was then used for inoculating 300 ml LB medium with 50 μg ampicillin/ml or 15 μg kanamycin/ml. When OD600 reached 0.7, 0.1 mM IPTG was added, and incubated in the presence of 0.1 g hemin/l at 16 °C for another 24 h. The cells were harvested by centrifugation at 5,000×g and 4 °C for 15 min.
The cell pellet was suspended in 15 ml lysis buffer (50 Mm Tris/HCl, pH 7.4, 150 mM NaCl, 1 % NP-40) with 1 mM PMSF as a protease inhibitor. The cell suspension was homogenized by a sonication at 0 °C. The cell lysate was then centrifuged at 20,000×g and 4°C for 25 min. Samples of supernatants and pellets were analyzed by SDS-PAGE and western blotting.
SDS-PAGE was performed in 12 % (v/v) polyacrylamide gels, using low protein molecular weight marker and Coomassie Blue R-250 staining. For western blotting, proteins were transferred from the gel onto a PVDF membrane. The membrane was blocked with 5% (w/v) skimmed milk in TBST (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 0.05 % Tween 20), incubated with the murine monoclonal anti-polyhistidine immunoglobulin G (IgG), rinsed three times with TBST, incubated with the goat anti-mouse IgG conjugated with alkaline phosphatase, rinsed three times with TBST, and detected with BCIP (5-bromo-4-chloro-3-indolyl phosphate)/nitro blue tetrazolium (NBT) solution.
All purification procedures were carried out at 4 °C. Nickel-chelating resin, 2 ml, was equilibrated with 5 column volumes of equilibration buffer (buffer A: 75 mM Tris/HCl, pH 8.0, 0.5 M NaCl). The crude supernatant was loaded onto the resin, which was sequentially washed with buffer A containing 20–250 mM imidazole. The enzyme purity was analyzed via SDS-PAGE. The fractions containing fusion VP were pooled, first dialyzed against 10 mM sodium tartrate (pH 5.5) supplemented with 1 mM CaCl2, then against 10 mM sodium tartrate (pH 5.5), finally concentrated with Amicon YM10 membrane (10 kDa cut-off). The protein concentration was determined by the Bradford method using bovine serum albumin as a standard.
The thioredoxin–hexahistidine tag was removed from fusion VP by enterokinase following the standard procedure. 8.07 mg fusion VP was incubated with 60 IU enterokinase in the cleavage buffer (25 mM Tris/HCl, pH 7.6, 50 mM NaCl, 2 mM CaCl2) at room temperature for 24 h, followed by a second nickel column. The flow-through was collected for further characterization.
Enzyme assay
Mn2+-independent oxidation of VA was estimated in 0.1 M sodium tartrate (pH 3.0) at 310 nm (veratraldehyde ε310 = 9,300 M−1 cm−1), oxidation of RB5 (ε598 = 30,000 M−1 cm−1) and ABTS (cation radical, ε418 = 36,000 M−1 cm−1) were followed in 0.1 M sodium tartrate (pH 3.5) at 598 and 418 nm, respectively (Ruiz-Dueňas et al. 2008; Garcia-Ruiz et al. 2012). Direct oxidation of Mn2+ was followed by the formation of the Mn3+-tartrate complex at 238 nm (ε238 = 6,500 M−1 cm−1) in 0.1 M sodium tartrate (pH 5.0) (Ruiz-Dueňas et al. 2008). All enzymatic assays were measured using initial rates taking linear increase or decrease (RB5) in the presence of 0.1 mM H2O2. The reactions were started by the addition of purified fusion VP and followed continuously. For determining specific activities against different substrates, 10 μM ABTS, 1 mM MnSO4, 10 mM VA, and 10 μM RB5 were used respectively. One unit of enzyme activity (U) is defined as the amount of the enzyme that catalyzes the conversion of 1 μmol substrate per min. K M and V max were determined using Lineweaver–Burk plot. k cat was calculated from V max on the basis of 52.6 kDa.
H2O2 stability assay
H2O2 stability was investigated by two approaches: (1) fusion VP activity against ABTS was measured in the presence of different H2O2 concentrations (0.01–6.4 mM) at 418 nm in 0.1 M sodium tartrate (pH 3.5). (2) Fusion VP (3 μM) solution was first incubated with 0.5, 1, and 2 mM H2O2 at 25 °C for different times (2–45 min) respectively, and then diluted to 0.1 mM H2O2. The residual activity was assayed using ABTS as substrate under same conditions as above.
Results and discussion
Cloning, expression and purification of fusion VP
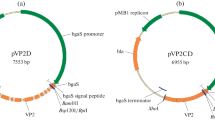
The codon optimized gene vpl2 encoding peroxidase VPL2 without its signal peptide was synthesized. The gene was cloned into vectors-pET-32a(+) and pET-28a(+) at restriction sites EcoRI and NotI, respectively (Fig. 1). The resulting constructs were named as pET32a-VP and pET28a-VP, respectively (Fig. 1). The pET32a-VP, including the VP nucleotide sequence fused after the coding sequences for the thioredoxin and hexa-histidine tags, was constructed since it has been suggested that thioredoxin could enhance the solubility of proteins normally produced in an insoluble form and stabilize the protein (Mohorčič et al. 2009). The pET28a-VP was constructed for expression of VP with the N-terminal His-tag.

Schematic structures of the expression vectors-pET28a-VP and pET32a-VP: ori the origin of replication, Kan kanamycin-resistance gene sequence, Amp ampicillin-resistance gene sequence, vp gene sequence encoding VP VPL2, lacI lacI repressor gene sequence, His-tag hexa-histidine tag, Trx-tag thioredoxin tag
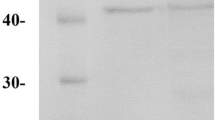
pET32a-VP was over-expressed in E. coli BL21(DE3) under IPTG induction in the presence of hemin. The optimized conditions for higher yield of soluble and active protein were 0.1 mM IPTG, 0.1 g hemin/L, and 16 °C for 24 h. SDS-PAGE (Fig. 2) and western blot (data not shown) analysis confirmed the presence of soluble fusion protein-THHVP (VP with the thioredoxin–hexahistidine tag). The predicted molecular weight for THHVP is 52.6 kDa, which corresponded to the SDS-PAGE value (Fig. 2). No major band corresponding to 52.6 kDa was observed in the vector-pET-32a(+) transformant (lane 11 in Fig. 2), neither detected by western blot (data not shown).

SDS-PAGE analysis of THHVP. Lane 1 low protein molecular weight marker, lane 2 crude supernatant for over-expressed THHVP, lane 3 flow-through when loaded, lane 4 eluent of buffer A, lanes 5–7 eluents of buffer A + 60 mM imidazole, lanes 8–10 eluents of buffer A + 100 mM imidazole, lane 11 crude supernatant for E. coli harboring the empty vector-pET-32a(+)
Under the same induction conditions pET28a-VP gave very low yield of protein based on SDS-PAGE and western blot (data not shown), which might be due to the absence of the thioredoxin tag in the expression vector-pET28a-VP (Fig. 1).
The over-expressed protein was purified by Ni-chelating affinity chromatography. The target protein was eluted with buffers containing 60 and 100 mM imidazole, with >85 % purity based on SDS-PAGE (Fig. 2). The collected fractions containing the target protein were combined, dialyzed, and concentrated. The yield of THHVP was about 12.5 mg/l.
The UV–Vis spectrum of THHVP showed a characteristic Soret peak at 407 nm (Fig. 3) demonstrating that hemin was incorporated into the heterologously over-expressed THHVP. The Rz value (A407/A280, a measure of hemin content of the peroxidases) for THHVP is 1.2, which is close to the values for fusion VPs from B. adusta expressed under auto-induction conditions (Mohorčič et al. 2009), but is much lower than 3.9 for the refolded non-fusion VP from the inclusion body (Pérez-Boada et al. 2002). The Rz value for VP (the thioredoxin–hexahistidine tag was removed from THHVP using enterokinase, there are nine amino acids left in the N-terminal.) is 2.5, indicating that the presence of the thioredoxin–hexahistidine tag has had some effects on the Rz value of THHVP. Between 400 and 750 nm, THHVP and VP showed three similar bands at 407 (Soret), 532, and 634 nm (Fig. 3), which means that the thioredoxin–hexahistidine tag had not affected the heme environment.

UV–Vis spectra of THHVP and VP (the thioredoxin–hexahistidine tag was removed using enterokinase). The enzymes were in 10 mM sodium tartrate (pH 5.5)

H2O2 dependence assay for THHVP. Enzyme activity against ABTS was assayed in the presence of different H2O2 concentrations (0.01–6.4 mM) at 418 nm in 0.1 M sodium tartrate (pH 3.5). The absolute value of 100 % relative activity is 0.17 ± 0.004 (ΔA418/min)
Enzymatic activity assay
Proteins expressed in E. coli lack adequate glycosylation and, although lack of glycosylation may not interfere with enzyme activity, its thermal stability can be lower than that of corresponding native enzyme (Nie et al. 1999). Thus, activity of VP over-expressed in E. coli should not be affected by lack of glycosylation.
Fusion protein THHVP oxidized all substrates tested: ABTS, Mn2+, VA, and RB5 (Table 1). Specific activities and kinetic parameters of non-fusion VP are also listed in Table 1. Specific activities against ABTS, Mn2+, VA, and RB5 were improved 9-, 13-, 18-, and 11-fold, respectively, by removing the thioredoxin–hexahistidine tag. K m were not affected by the tag whereas the turnover numbers were. The catalytic efficiencies (k cat /K m) against ABTS, Mn2+, VA, and RB5 increased 12-, 11-, 6-, and 12-times, respectively, after the tag was removed. These data have demonstrated that the tag has greatly affected enzyme activity.
H2O2 stability study
Peroxidases tend to lose activity in the presence of H2O2 through a mechanism-based process known as a suicide inactivation (Valderrama et al. 2002). The optimum H2O2 concentration for THHVP is 0.1 mM, above which enzyme gradually lost activity, it retained only about 4.6 % of original activity when H2O2 reached 6.4 mM (Fig. 4). THHVP lost 50 % of original activity after being incubated with 0.5, 1, and 2 mM H2O2 for 25, 6, and 1 min, respectively (Fig. 5) and completely lost activity with 2 mM H2O2 for 30 min (data not shown). In addition, inactivation of VP by H2O2 is dependent on pH (Böckle et al. 1999). Thus, protein engineering has to be carried out for VP in order to improve its H2O2 stability (Garcia-Ruiz et al. 2012).

H2O2 stability assay for THHVP. THHVP (3 μM) solution was first incubated with 1 mM H2O2 at 25 °C for different times (2–45 min), then diluted to 0.1 mM H2O2. The residual enzyme activity was measured using ABTS as substrate under standard conditions. The absolute value of 100 % residual activity is 0.1 ± 0.005 (ΔA418/min)
Conclusion
Active VP from P. eryngii was directly produced in E. coli under IPTG induction in the presence of hemin at moderate yields, thus the labor-intensive, time-consuming, and low-efficient refolding was avoided. This will be useful for production of active VP for structure–function investigation.
References
Böckle B, Martínez MJ, Guillén F, Martínez AT (1999) Mechanism of peroxidase inactivation in liquid cultures of the ligninolytic fungus Pleurotus pulmonarius. Appl Environ Microbiol 65:923–928
Doyle WA, Smith AT (1996) Expression of lignin peroxidase H8 in Escherichia coli: folding and activation of the recombinant enzyme with Ca2+ and haem. Biochem J 315:15–19
Garcia-Ruiz E, Gonzalez-Perez D, Ruiz-Dueňas FJ, Martínez ÁT, Alcalde M (2012) Directed evolution of a temperature-, peroxide- and alkaline pH tolerant versatile peroxidase. Biochem J 441:487–498
Lu-Chau TA, Ruiz-Duenas FJ, Camarero S, Feijoo G, Martinez MJ, Lema JM, Martinez AT (2004) Effect of pH on the stability of Pleurotus eryngii versatile peroxidase during heterologous production in Emericella nidulans. Bioprocess Biosyst Eng 26:287–293
Martínez MJ, Ruiz-Dueňas FJ, Guillén F, Martínez ÁT (1996) Purification and catalytic properties of two manganese peroxidase isoenzymes from Pleurotus eryngii. Eur J Biochem 237:424–432
Martínez ÁT, Ruiz-Duenas FJ, Martínez MJ, Río JC, Gutiérrez A (2009) Enzymatic delignification of plant cell wall: from nature to mill. Curr Opin Biotechnol 20:348–357
Mayfield MB, Kishi K, Alic M, Gold MH (1994) Homologous expression of recombinant manganese peroxidase in Phanerochaete chrysosporium. Appl Environ Microb 60:4303–4309
Mester T, Field JA (1998) Characterization of a novel manganese peroxidase-lignin peroxidase hybrid isozyme produced by Bjerkandera species strain BOS55 in the absence of manganese. J Biol Chem 273:15412–15417
Mohorčič M, Benčina M, Friedrich J, Jerala R (2009) Expression of soluble versatile peroxidase of Bjerkandera adusta in Escherichia coli. Bioresour Technol 100:851–858
Nie GJ, Reading NS, Aust SD (1999) Relative stability of recombinant versus native peroxidases from Phanerochaete chrysosporium. Arch Biochem Biophys 365:328–334
Pease EA, Aust SD, Tien M (1991) Heterologous expression of active manganese peroxidase from Phanerochaete chrysosporium using the baculovirus expression system. Biochem Biophys Res Commun 179:897–903
Pérez-Boada M, Doyle WA, Ruiz-Dueňas FJ, Martínez MJ, Martínez AT, Smith ÁT (2002) Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzyme Microb Tech 30:518–524
Pérez-Boada M, Ruiz-Dueňas FJ, Pogni R, Basosi R, Choinowski T, Martínez MJ, Piontek K, Martínez ÁT (2005) Versatile peroxidase oxidation of high potential aromatic compounds: site-directed mutagenesis, spectroscopic and crystallographic investigation of three long-range electron transfer pathways. J Mol Biol 345:385–402
Ruiz-Dueňas FJ, Morales M, Mate MJ, Romero A, Martínez MJ, Smith AT, Martínez ÁT (2008) Site-directed mutagenesis of the catalytic tryptophan environment in Pleurotus eryngii versatile peroxidase. Biochemistry 47:1685–1695
Valderrama B, Ayala M, Vazquez-Duhalt R (2002) Suicide inactivation of peroxidases and the challenge of engineering more robust enzymes. Chem Biol 9:555–565
Whitwam RE, Gazarian IG, Tien M (1995) Expression of fungal Mn peroxidase in E. coli and refolding to yield active enzyme. Biochem Biophys Res Commun 216:1013–1017
Acknowledgments
We are grateful to Knowledge Innovation Program of the Chinese Academy of Sciences (Grant KSCX1-YW-11A3) and Natural Science Foundation of Shandong Province (China) (Grant ZR2010CM058) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bao, X., Liu, A., Lu, X. et al. Direct over-expression, characterization and H2O2 stability study of active Pleurotus eryngii versatile peroxidase in Escherichia coli . Biotechnol Lett 34, 1537–1543 (2012). https://doi.org/10.1007/s10529-012-0940-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-012-0940-5