
Abstract
Hydroxyapatites were analysed using electron microscopy, X-ray diffraction (XRD) and X-ray fluorescence (XRF) analysis. Examination of a bacterially produced hydroxyapatite (Bio-HA) by scanning electron microscopy showed agglomerated nano-sized particles; XRD analysis confirmed that the Bio-HA was hydroxyapatite, with an organic matter content of 7.6%; XRF analysis gave a Ca/P ratio of 1.55, also indicative of HA. The size of the Bio-HA crystals was calculated as ~25 nm from XRD data using the Scherrer equation, whereas Comm-HA powder size was measured as ≤50 μm. The nano-crystalline Bio-HA was ~7 times more efficient in removing Sr2+ from synthetic groundwater than Comm-HA. Dissolution of HA as indicated by the release of phosphate into the solution phase was higher in the Comm-HA than the Bio-HA, indicating a more stable biomaterial which has a potential for the remediation of contaminated sites.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Current technologies for the removal of radioactive metals (i.e. U, Tc, Sr, Co, Pu) from nuclear waste waters include osmosis, ultrafiltration and precipitation (Cecille et al. 1991). These technologies tend to be expensive and require high maintenance. Another approach is to use a sorbent or ion exchange material that can be disposed of once its full capacity is reached. Traditional ion exchangers such as titanates and hexacyanoferrate are standard in the nuclear industry but these can be rather specific for particular target metals (IAEA 2002), whereas recently-described biogenic phosphate mineral matrices based on uranyl hydrogen phosphate (HUP; Paterson-Beedle and Macaskie 2006) and zirconium phosphate (Zr–P; Mennan et al. 2010) take up a range of metals and have the potential to operate under a wide range of pH values, with a potential capacity several orders of magnitude larger than traditional materials (Mennan et al. 2010). Furthermore, growth of this biomass is rapid and cheap (several kg fresh weight after 24 h in batch growth at 30°C; Macaskie et al. 1995); biogenic material does not require procurement of materials (e.g. natural zeolites) from mined sources, and gives the potential for rapid manufacture of portable treatment units in response to a sudden contamination incident.
With projected new nuclear reactor builds and operations worldwide underway in response to the urgent need for low carbon energy technologies, there is a pressing need for new treatment technologies to manage historic wastes effectively while also providing a robust foundation for the management of future wastes.
Hydroxyapatite [HA; Ca5(PO4)3(OH)] is the main inorganic component of bones and teeth. This material has previously been found to be efficient in the removal of radioactive ions such as UO2 2+ (Simon et al. 2008), Co2+ (Smiciklas et al. 2006) and Sr2+ (Thomson et al. 2003) from solution. The exact mechanism of divalent metal ion removal (M2+) is not fully understood and the mechanism may vary according to the metal used (Simon et al. 2008). For Sr2+ the metal ion may sorb to the HA surface according to Eqs. 1 and 2 or undergo ion exchange (3).
In a similar way to the synthesis of HUP and Zr–P (above), a Serratia sp. was found to biomanufacture nanophase hydroxyapatite as a potential bone replacement material (Thackray et al. 2004; Ledo et al. 2008) from glycerol 2-phosphate (G2P) and Ca2+. The G2P is cleaved via an atypical phosphatase enzyme present at high levels in appropriately-grown cells, and liberated phosphate precipitates with M2+ on the bacterial cell surface (Thackray et al. 2004). This nano-sized HA material may have great potential to remove radioactive waste metals due its larger and more reactive surface area.
In an aqueous environment HA can undergo dissolution, releasing Ca2+, PO4 3− and OH− ions into solution according to Eq. 4. The rate of dissolution may vary according to the pH and temperature of the solution matrix, and the exchange reactions of Sr2+ for soluble Ca2+ may occur at equilibrium, limiting the long-term efficacy of HA-materials for radionuclide immobilisation. Thus, in addition to an ability to sorb radionuclides, the ability to resist dissolution reactions is very important.
The objective of this study was to evaluate the potential of biogenic hydroxyapatite (Bio-HA) made under similar conditions as used for the preparation of bone substitute material (Ledo et al. 2008) as a novel substrate for the remediation of radionuclides. To this end, two factors need to be considered: the efficacy of radionuclide uptake, and the stability of the material in aqueous conditions. This study investigated the uptake ability of Bio-HA for the removal of aqueous Sr2+ ions from synthetic groundwater, comparing its efficacy against commercially produced hydroxyapatite (Comm-HA). We also compared the ability of the two HAs to liberate phosphate and calcium ions into the groundwater solution matrix (dissolution reactions).
Materials and methods
Preparation of biomass
Serratia sp. NCIMB 40259 was used by kind permission of Isis Innovation, Oxford, UK. Cells were grown in carbon (lactose)-limited continuous culture at 30°C as described previously (Finlay et al. 1999). Minimal medium (2.5 l) was added to an air lift fermenter containing up to 200 polyurethane reticulated foam cubes (1 cm3; TM30 supplied by Recticel, Wetteren, Belgium) threaded on cotton. The culture was started by the addition of 50 ml inoculum which had been pre-grown in similar medium. The culture was allowed to grow batch-wise, switched to continuous mode after 24 h (OD600 ~ 0.6) and maintained at steady-state for 6 days. The culture was monitored by measurement of phosphatase activities and cell densities of free cells in the culture outflow (Finlay et al. 1999). The foam cubes (heavily loaded with biofilm) were withdrawn and stored suspended from the top of a closed vessel over isotonic saline (8.5 g/l; to catch any shed biofilm) at 4°C until use.
Phosphatase activity assay
Phosphatase activity was assayed by the release of p-nitrophenol (PNP) from p-nitrophenyl phosphate (PNPP) (Finlay et al. 1999). Phosphatase activity (units) is defined as nmol of PNP liberated/min per mg bacterial protein. The OD600 value of the cells was converted to bacterial protein using a conversion factor obtained by the Lowry assay (0.278 mg protein/ml at OD600 = 1; path length = 1 cm).
Bioaccumulation of calcium phosphate by Serratia sp.
The influence of sodium glycerol 2-phosphate (G2P) concentration on the rate of biomineralisation was investigated (two replicate experiments) in order to determine whether an excess of G2P was required for maximum HA accumulation. Ten biofilm-loaded foam cubes (prewashed in isotonic saline; ~4.5 mg dry cell mass/cube) were placed in flasks containing 50 ml TAPSO/NaOH buffer (50 mM; pH 9.2) with either 1 mM (n = 3) or 5 mM (n = 3) G2P and 1 mM CaCl2. Control flasks were prepared in the same way without G2P; all flasks were shaken gently at room temperature. After 24 h the flasks were sub-sampled daily to monitor Ca2+ and phosphate concentrations. The volume reduction was replaced daily with additional G2P (to either 1 mM or 5 mM additional amounts) and CaCl2 (to 1 mM additional amount), so that the total amount of Ca2+ added after 5 days was 5 mM and G2P was either 5 mM or 25 mM in these initial tests. For preparation of material for solid state analysis and subsequent determination of Sr2+ uptake the suspension volume for 10 cubes was increased to 100 ml (preliminary studies; daily dosing as above for 8 days) and then to 1 l with all ingredients scaled up accordingly. The harvest from the original culture vessel (200 cubes) was divided. A portion of cubes was used for another study investigating the preparation of bone precursor material via sintering to destroy bacteria and potential pyrogens (Ledo et al. 2008). The remaining cubes (100 each) from three independent fermenters (3 × 100 cubes) were used in the preparation of material (bio-hydroxyapatite; ‘Bio-HA’; 3 × 1000 ml suspensions) for this study, and the material was pooled to obtain sufficient quantity for all the tests.
Assay of phosphate and calcium during uptake and HA manufacture onto biomass
Samples (≤20 mM phosphate) were diluted 30-fold. Sodium molybdate (0.6 ml; 25 g/l in 1.67 M H2SO4) was added to 1 ml of sample. Phosphate concentrations were determined after the addition of SnCl2 (0.4 ml; 1.5 g/l in 1 M HCl). The blue complex was measured at A720 nm and the phosphate concentration determined by reference to a standard phosphate curve.
For the determination of Ca2+, a sample (0.1 ml; ≤1 mM) was added to MOPS/NaOH buffer (1.9 ml; 20 mM; pH 7). Calcium concentrations were determined after the addition of Arsenazo III solution (0.1 ml; 1.5 g/l). The pink complex was measured at 631 nm and the Ca2+ concentration determined by reference to a standard Ca2+ curve.
Sampling and characterisation of hydroxyapatites
After 8 days the foam cubes containing calcium phosphate-loaded Serratia sp.cells were harvested. The material was dislodged and collected from the foam cubes by a gentle tapping and squeezing action. A portion of freshly harvested material was characterised by scanning electron microscopy (SEM) and X-ray powder diffraction (XRD), the remaining material was washed in acetone, air dried and stored at room temperature for ~4 years; this stored material is referred to as Bio-HA. For comparison a reagent grade commercially available hydroxyapatite powder (Comm-HA; Sigma–Aldrich; Part number: 289396) was obtained. A previous study used ‘Capital 60’ hydroxylapatite (Plasma Biotal Ltd, Derbyshire UK) with a particle size determined as 167 nm (Yong et al. 2003). In this study the Sigma–Aldrich material was used due to its smaller particle size, i.e. closer to the bacterially-manufactured material (see later). The Comm-HA and Bio-HA were characterised using an environmental SEM, XRD and X-ray fluorescence (XRF).
Scanning electron microscope analysis
Freshly harvested material containing calcium phosphate-loaded Serratia sp. cells and Serratia sp. cells without calcium phosphate were fixed for 1 h using 2.5% glutaradehyde (in 0.1 M sodium cacodylate/HCl buffer pH 5.2) at 4°C. The cells were dehydrated at room temperature and imaged by SEM as described previously (Yong et al. 2002). Samples of Bio-HA and Comm-HA were mounted onto stubs, platinum-coated and imaged under high vacuum mode using an environmental SEM (FEI Philips FEG ESEM XL30; Detector 15 kV).
X-ray fluorescence
The elemental composition of Bio-HA and Comm-HA were determined (wt% of each element) using X-ray fluorescence (XRF; Bruker S8). An accurately weighed mass (0.2 g) of powdered sample was analysed using an Rh X-ray source and helium atmosphere. The Ca/P molar ratios were determined by the same instrument using (5 point) calibrated hydroxyapatite standards (Sigma–Aldrich) prepared in Hoechst wax.
X-ray powder diffraction analysis
A portion of harvested air dried material containing calcium phosphate-loaded Serratia sp. cells was analysed by XRD as described previously (Yong et al. 2002). Samples of Bio-HA and Comm-HA were analysed by XRD (Bruker D8 Advanced X-ray diffractometer; 10–60°; (2θ; step 0.009°; count time 0.61 s; Cu Kα radiation)). The particle size of Bio-HA was calculated from diffraction patterns using the Scherrer equation (Patterson 1939).
Loss of organic natter by ignition
A portion of accurately weighed Bio-HA material (~200 mg) was placed in a furnace-cleaned (600°C, 2 h) ceramic boat. To prevent loss of material due to sputtering, the Bio-HA was covered (5 mm) with glass wool, accurately weighed and heated at 600°C for 2 h. The material was allowed to cool in a desiccator and reweighed to determine % organic matter.
Uptake of Sr2+ from aqueous solutions
Bio-HA and Comm-HA were tested for the removal of Sr2+ from solution, using a non-radioactive isotope (89Sr). A Sr2+ solution was prepared in artificial ground water (Table 1) using high purity water (MQ water, ≥18.2 MΩ/cm) to give 100 mg Sr/l (1.14 mM).
All sorption experiments were carried out in triplicate. Accurately weighed masses (~0.025 g) of Bio-HA (n = 24) and Comm-HA (n = 24) were placed in a polypropylene vials (Eppendorf). An aliquot (1.5 ml) of Sr2+ solution was pipetted into the vials, the vials were then immediately positioned vertically on an orbital shaker (150 rpm) at 10°C until time of analysis. At intervals, samples from three vials were harvested by centrifugation (10,000×g) and the supernatant analysed by ion chromatography to determine the solution phase concentrations of Sr2+, Ca2+ (Dionex ICS-1100) and PO4 3− (Dionex ICS-90).
Results and discussion
Removal of calcium from solution supported by G2P hydrolysis, and HA biomanufacture
The phosphatase specific activity of the cells at harvest, measured at pH 7.0, was 2384 U. A more detailed study of the immobilised cells and their bio-HA coating as examined by a variety of non-invasive methods was described previously (Macaskie et al. 2005). Preliminary tests established that by using a pH of 9.2 (optimal for alkaline earth metal phosphate precipitation as shown in preliminary studies) the phosphatase activity was reduced by ~35% but was still sufficiently high to obtain effective calcium phosphate mineralisation. Initial tests using 1 mM G2P and 1 mM Ca2+ showed that significant removal of Ca2+ did not occur until after 2 days (Fig. 1a) and increasing the G2P concentration to 5 mM reduced this delay to 1 day (Fig. 1b). After 5 days ~5 mM Ca2+ was removed with ~20 mM of residual phosphate in solution (Fig. 1b). Precipitation continued over longer periods of incubation (up to and beyond 8 days), with complete removal of Ca2+ each day from the 50 and 100 ml suspensions. Negligible Ca2+ was removed by cell suspensions without G2P. Assuming that all cells participated equally (permeation of solution to all areas of the biofilm within the cubes was shown previously by magnetic resonance imaging: Macaskie et al. 2005) the total biomass available was ~45 mg (10 cubes) and hence by calculation 16 mg of Ca2+ was removed in the 50 ml flasks and, by extrapolation, ~320 mg Ca2+/l, i.e. ~1 g of total Ca2+ (from 3 l) for the subsequent tests.

Assay of phosphate and calcium during uptake and HA manufacture onto biomass. Ten biofilm-foam cubes (~4.5 mg dry cell mass per cube; phosphatase activity was 2,834 units at harvest) were challenged with 50 ml TAPSO/NaOH buffer (50 mM; pH 9.2), and either a 1 mM CaCl2; 1 mM G2P or b 1 mM CaCl2; 5 mM G2P. Appropriate Ca (to 1 mM added) and G2P (to 1 mM or 5 mM added) concentrations were added daily. a, b Residual phosphate (filled circle) and removed Ca2+ (filled square) by Serratia sp. cells
Examination of the cells in suspensions unsupplemented with G2P showed no accumulated biominerals (Fig. 2a) whereas the Ca2+ and G2P-challenged bacteria were embedded in a mass of crystalline material with occasional ‘naked’ cells (which presumably arose from deeper biofilm layers: Fig. 2b). Use of biofilm-derived cells was important for comparison with the previous bone-precursor material studies (Ledo et al. 2008) which gave a full characterisation of the Bio-HA at the time of its manufacture. Immobilisation of Bio-HA onto the polyurethane reticulated foam matrix is ideal for the production of a bone precursor material since the foam and bacteria are destroyed on sintering to leave a skeleton of HA mineral (Ledo et al. 2008) which is very conducive to bone cell ingress (Sammons et al. 2007). Of greater potential importance in the current study is the potential contribution of the extracellular polymeric material (EPM) to HA scaffolding and its role in the stabilisation of nanoparticles. Once formed the HA requires no further enzymatic activity and hence dried material could have a potentially long shelf-life, akin to other inorganic ion-exchangers.

Calcium phosphate accumulation by Serratia cells viewed under SEM. Freshly harvested material air-dried prior to examination (see text), showing a Serratia sp.cells and b material containing calcium phosphate-loaded Serratia sp. cells; individual cells are clearly visible within the material (arrowed in B). Bars shown are 10 μm
X-ray fluorescence of hydroxyapatite
The wt% elemental composition of Bio-HA and Comm-HA are shown in Table 2. The Bio-HA contains some low level metal impurities (i.e. Na, K, S, Mg, Zn, Cu, Sr), which may be attributed to traces supplied by the chemical reagents or biomass trace elements (Zn, Cu).
The Ca/P molar ratios are also shown in Table 2. The Ca/P ratio of Bio-HA was calculated as 1.55/1; this is in close agreement with that of Ledo et al. (2008) at 1.62/1. Although theoretical HA [(Ca5(PO4)3(OH)] has a Ca/P molar ratio of 1.67/1, the molar ratio can vary between 1.5/1 and 1.71/1 depending on the preparation method (Benhayoune et al. 2001). The Ca/P ratio of Comm-HA and Bio-HA are similar at 1.58/1 and 1.55/1, respectively. The Bio-HA is slightly more Ca and P deficient than the Comm-HA (Table 2), this may be attributed to the Bio-HA containing organic (i.e. residual Serratia cells) and biomass impurities.
Scanning electron microscopy (SEM) of stored Bio-HA
Examination of materials by scanning electron microscopy shows that the Comm-HA product (Fig. 3a, b) comprises heterogeneous spherical particles of varying sizes (≤50 μm). The Bio-HA (Fig. 3c, d) appears to consist of agglomerated nanoparticles; it was therefore not possible to calculate the particle size from SEM. At higher resolution the surface of the Comm-HA appears irregular (Fig. 3b) and the Bio-HA appears as discrete ‘nodules’ with, in some cases, cell ‘ghosts’ apparent (Fig. 3d, arrowed).

SEM Images of hydroxyapatite. a, b Comm-HA and c, d Bio-HA. Bars shown are 50 μm (a, b) and 5 μm (c, d). The material was acetone-washed but cell ‘ghosts’ are visible (arrowed in D)
X-ray powder diffraction (XRD) analysis
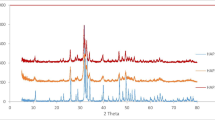
X-ray powder patterns for the Bio-HA and Comm-HA are shown in Fig. 4. The peaks were matched against calcium hydrogen phosphate hydroxide (JCPDS database; Pattern number 00-046-0905) indicating a hydroxyapatite material. The less well defined peaks of the Bio-HA are indicative of a lower crystallinity, i.e. nanomaterial form. Accordingly, the particle size of the Bio-HA was calculated as ~25 nm using the Scherrer equation. This is in close agreement with the earlier result using the freshly harvested HA material (Ledo et al. (2008) and identical powder patterns were obtained with stored material, showing complete stability of the material on long term storage (~4 years) at room temperature in air.

X-ray powder diffraction patterns of Bio-HA and Comm-HA. Peaks matched against calcium phosphate hydroxide (PDF database; Pattern number 00-046-0905)
Loss of organic matter by ignition
Organic matter was calculated from loss by ignition as 7.6% and is attributed to Serratia cells and/or extracellular polymeric material produced during batch methods. Ghost cells have been imaged in freshly harvested material Fig. 3d.
Uptake of Sr2+ from artificial groundwater and dissolution of the minerals in groundwater
Uptake of Sr2+ by Bio-HA and Comm-HA was compared. Uptake of Sr2+ by Bio-HA (Fig. 5a) was very efficient. Within 30 min there was a rapid decrease in the amount of aqueous Sr2+ from the initial concentration of 1.14 ± 0.01 to 0.29 ± 0.01 mM. The concentration of aqueous Sr2+ then fluctuated (±0.05 mM) and decreased to 0.2 ± 0.01 mM at 25 h. In comparison, the uptake of Sr2+ by Comm-HA was less efficient (Fig. 5a). There was only a small decrease in aqueous Sr2+, from the initial concentration of 1.14 ± 0.01 to 1.05 ± 0.001 mM, in 30 min. Aqueous Sr2+ then slowly decreased to give a final concentration of 1.00 ± 0.003 mM at 25 h. At the end of the experimental period (25 h) Comm-HA had taken up 0.76 mg Sr2+/g whereas the Bio-HA had immobilised 5.35 mg Sr2+/g of HA material.

Changes in solution chemistry for suspensions containing Comm-HA (filled square) and Bio-HA (filled circle). Times symbol highlights initial (0 h) solution concentration a Sr2+ uptake, b Ca2+ and c PO4 3− release; and d the correlation between Sr2+ uptake and Ca2+ release for Comm-HA. Error bars show ±1SD (n = 3)
Calcium uptake was also observed by Bio-HA (Fig. 5b) with initial groundwater concentrations decreasing from 1.4 ± 0.07 to 1.24 ± 0.003 mM within 30 min, the Ca2+ concentrations then fluctuated (±0.1 mM) to give 1.35 ± 0.04 mM at 25 h. The Comm-HA showed an increase in solution phase Ca2+ (Fig. 5b) from the initial groundwater concentration of 1.40 ± 0.07 to 1.44 ± 0.002 mM within 30 min; the Ca2+ concentrations then fluctuated (±0.1 mM) to give final concentration of 1.47 ± 0.004 mM.
Phosphate increase in the solution phase was observed in both the Comm-HA and Bio-HA experiments (Fig. 5c), suggesting dissolution of HA (Eq. 4). Phosphate was first detected in the solution phase of the Comm-HA from 1 h (0.01 ± 0.01 mM) and the concentration steadily increased to 0.17 ± 0.03 mM at 25 h. Phosphate in the solution phase of the Bio-HA was first observed at 6 h (0.01 ± 0.01 mM) and the concentration increased to 0.13 ± 0.02 mM at 25 h. The lower amount of PO4 3− in the solution for Bio-HA may indicate less dissolution of HA (Eq. 4) and hence a more stable material in a groundwater solution matrix.
Only the Comm-HA showed a linear (0.84 R 2) correlation between Sr2+ uptake and Ca2+ release (Fig. 5d). The gradient of the trend line (1.33) indicates a close to 1:1 ion exchange of Sr2+ for Ca2+ (Eq. 3); ion exchange has been suggested previously for the uptake of Sr2+ by hydroxyapatite (Lazić and Vuković 1992). The Bio-HA showed uptake of Ca2+ and Sr2+ from groundwater and did not appear to show the same ion exchange behaviour as Comm-HA. The increased efficiency of the Bio-HA may be due to its increased surface area of the nano-crystals (~25 nm).
Conclusions
This study shows that nano-crystalline Bio-HA (~25 nm) is 7 times more effective than commercial HA (≤50 μm) for the uptake of aqueous Sr2+, accumulating 5.35 mg/g Bio-HA material in 25 h, compared to 0.76 mg/g Comm-HA. Comm-HA showed a near 1:1 ion exchange of Sr2+ for Ca2+, but the mechanism of Sr2+ accumulation by Bio-HA appears more complex with both Sr2+ and Ca2+ uptake occurring. This biologically-produced nano-sized HA material has the potential to be used for radionuclide cleanup technologies; current studies are characterizing Bio-HA in detail and investigating uptake of other problematic radionuclide contaminants, including Co and U. More extensive studies using real groundwater will be reported in full in a later publication.
References
Benhayoune H, Charlier D, Jallot E, Laquerriere P, Balossier G, Bonhomme P (2001) Evaluation of the Ca/P concentration ratio in hydroxyapatite by STEM–EDXS influence of the electron irradiation dose and temperature processing. J Phys D Appl Phys 34:141–147
Cecille L, Casarci M, Pietrelli L (1991) New separation chemistry techniques for radioactive waste and other specific applications. University Press, Cambridge
Finlay JA, Allan VJM, Conner A, Callow ME, Basnakova G, Macaskie LE (1999) Phosphate release and heavy metal accumulation by biofilm-immobilized and chemically-coupled cells of a Citrobacter sp. pre-grown in continuous culture. Biotechnol Bioeng 63:87–97
IAEA (2002) Application of ion exchange processes for the treatment of radioactive waste and management of spent ion exchangers. Technical report series No. 408. International Atomic Energy Agency (IAEA), Vienna
Langford JI (1992) Proceedings of the international conference on accuracy in powder diffraction II. NIST special publications 846, pp 110–126
Lazić S, Vuković Ž (1992) Ion exchange of strontium on synthetic hydroxyapatite. J Radioanal Nucl Chem 149:161–168
Ledo HM, Thackray AC, Jones IP, Marquis PM, Macaskie LE, Sammons RL (2008) Microstructure and composition of biologically synthesized hydroxyapatite. J Mater Sci Mater Med 19:3419–3427
Macaskie LE, Hewitt CJ, Shearer JA, Kent CA (1995) Biomass production for the removal of heavy metals from aqueous solution at low pH using growth-decoupled cells of a Citrobacter sp. Int Biodeterior Biodegrad 35:73–92
Macaskie LE, Yong P, Paterson-Beedle M, Thackray AC, Marquis PM, Sammons RL, Nott KP, Hall LD (2005) Novel non line of-sight method for coating hydroxyapatite onto surfaces of support materials by biomineralization. J Biotechnol 118:187–200
Mennan C, Paterson-Beedle M, Macaskie LE (2010) Accumulation of zirconium phosphate by a Serratia sp.: a benign system for the removal of radionuclides from solution. Biotechnol Lett (in press)
Paterson-Beedle M, Macaskie LE (2006) Utilisation of a hydrogen uranyl phosphate-based ion exchanger supported on a biofilm for the removal of cobalt, strontium and caesium from aqueous solutions. Hydrometallurgy 83:141–145
Patterson AL (1939) The Scherrer formula for X-ray particle size determination. Phys Rev 56:978–982
Rahman H (2006) Virus–colloid interactions in groundwater. PhD thesis, University of Birmingham, UK
Sammons RL, Thackray AC, Ledo HM, Marquis PM, Jones IP, Yong P, Macaskie LE (2007) Characterisation and sintering of nanophase hydroxyapatite synthesised by a species of Serratia. J Phys Conf Ser 93:4–7
Simon FG, Biermann V, Peplinski B (2008) Uranium removal from groundwater using hydroxyapatite. Appl Geochem 23:2137–2145
Smiciklas I, Dimovic S, Plecas I, Mitric M (2006) Removal of Co2+ from aqueous solutions by hydroxyapatite. Water Res 40:2267–2274
Thackray AC, Sammons RL, Macaskie LE, Yong P, Marquis PM (2004) Bacterial biosynthesis of calcium bone-substitute material. J Mater Sci Mater Med 15:403–406
Thomson BM, Smith CL, Busch RD, Siegel MD, Baldwin C (2003) Removal of metals and radionuclides using apatite and other natural sorbents. J Environ Eng ASCE 129:492–499
Yong P, Rowson NA, Farr JPG, Harris IR, Macaskie LE (2002) Bioaccumulation of palladium by Desulfovibrio desulfuricans. J Chem Technol Biotechnol 77:593–601
Yong P, Sammons RL, Marquis PM, Lugg H, Macaskie LE (2003) Synthesis of nanophase hydroxypatite by Serratia sp. N14. In: Tsezos M, Hatzikioseyian A, Remoundaki E (eds) Biohydrometallurgy: a sustainable technology in evolution. National Technical University of Athens, Athens, pp 1205–1214. ISBN 960-88415-0-X
Acknowledgments
This work was supported by the EPSRC (grants EP/E012213/1 and EP/G063699/1). The authors thank Dr. Louise Male and Mr. Colin Slater for supporting XRD and XRF analysis, and Professor Keiko Sasaki and Ms. Sayo Moriyama for useful discussions. The latter collaboration was supported by the BBSRC (Japan Partnering Award).
Author information
Authors and Affiliations
Corresponding author
Additional information
Purpose of work Strontium (90Sr) is an important component of radioactive waste produced from the fission of 235U. Hydroxyapatites (HA) have been previously investigated for the removal of fission products. Nano-crystalline, bacterially produced hydroxyapatite (Bio-HA) has now been evaluated against a commercial HA (Comm-HA) for the sorption and immobilisation of aqueous Sr2+ ions.
Rights and permissions
About this article
Cite this article
Handley-Sidhu, S., Renshaw, J.C., Yong, P. et al. Nano-crystalline hydroxyapatite bio-mineral for the treatment of strontium from aqueous solutions. Biotechnol Lett 33, 79–87 (2011). https://doi.org/10.1007/s10529-010-0391-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-010-0391-9