
Abstract
An improved transformation method for the biocontrol agent, Beauveria bassiana, was developed. For convenience of transformation selection and detection, the coding regions of the genes for phosphinothricin acetlytransferase and green fluorescent protein were fused and an expression vector, pBFT, carrying this fusion was constructed. Under optimum conditions, over 60 transformants μg−1 plasmid DNA were obtained. B. bassiana conidia frozen 1 month at −80°C were fully competent for transformation. The method was significantly less laborious and more rapid than current methods for B. bassiana. The bar::egfp provides a selectable and visible marker which may expedite future genetic engineering of this fungus.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The entomopathogenic fungus, Beauveria bassiana, is an important biological control agent for a variety of insect pests, including both agricultural pests and vectors of human pathogens. In recent years, B. bassiana research has received greater attention and efforts to determine the molecular basis of its development, virulence and pathogenicity have expanded (Cho et al. 2006a, b; Fan et al. 2007; Fang et al. 2005). To produce transgenic B. bassiana, several genetic transformation methods including protoplast-based, blastospore-based and Agrobacterium-mediated methods, have been developed. Protoplast-based methods (Daboussi et al. 1989; Pfeifer and Khachatourians 1992) are time-consuming and have low efficiency. The blastospore-based method is also time-consuming, requiring 3 d to prepare blastospores (Jiang et al. 2007; Ying and Feng 2006). The higher efficiency of the Agrobacterium-mediated transformation method is attractive; however, target gene cassettes must be constructed in a binary vector and mobilized into Agrobacterium strains prior to B. bassiana transformation and co-cultivation of the fungus and bacterium requires 48 h (Fang et al. 2004). Fungal conidia have been successfully employed as competent cells in the electroporation transformation of several filamentous fungi (Robinson and Sharon 1999; Sánchez and Aguirre 1996). Germinated conidia have proved to be most competent for receiving foreign DNA before nuclear division and septa formation in Colletotrichum gloeosporioides (Robinson and Sharon 1999) and Aspergillus nidulans (Koukaki et al. 2003).
Here we describe an improved method for genetically transforming B. bassiana with pBFT, a new expression vector that we constructed. The vector contains the marker genes for phosphinothricin acetlytransferase and green fluorescence protein as a fusion sequence, bar::egfp. We demonstrate that the method is simple, reliable, and highly efficient, and thus potentially very useful for genetic research with B. bassiana.
Materials and methods
Strains and culture conditions
Beauveria bassiana strain Bb0062 single spore isolate and cultivation were previously described (Fang et al. 2005).
Construction of the vector of fusion gene bar::egfp
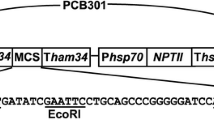
For convenience of transformation selection and detection, a plasmid containing the fusion gene bar::egfp was constructed (Fig. 1). The bar and egfp were amplified from PCB200 (Fang et al. 2004) and pEGFP-C1 (Clontech) using primers B-F/B-R and G-F/G-R (Table 1), respectively. All the amplified fragments were cloned into pUCm-T (Sangon), and sequenced. The bar was excised from the cloning vector by NotI and XbaI, and fused upstream to the egfp to form pUC-bar::egfp. Then, the fusion gene was linked with PgpdA promoter and trpC terminator from Aspergillus nidulans in plasmid pUC-PT to form the final plasmid, designated as pBFT.

Design of the transformation plasmid pBFT. The fusion gene bar::egfp was under the control of the A. nidulans gpd promoter (PgpdA); efficient termination was facilitated by the A. nidulans trpC terminator. The cleavage sites for the endonucleases EcoRI, NotI, BamHI, XbaI and HindIII were indicated
Preparation of competent cells
The conidia were cultivated in SDY at 26°C on a rotary shaker at 180 rpm. To determine the germination stage of conidia, the cultures were observed at 0, 8, 12, and 16 h after inoculation. To confirm the nucleus number in the germinated conidia, the conidia were stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) and imaged in the same microscopic field of light and fluorescence.
To prepare competent cells, 500 ml SDY medium was inoculated with approximately 5 × 109 conidia and incubated at 26°C on a rotary shaker at 180 rpm. Aliquots of 50 ml (about 5 × 108 conidia) were rapidly removed at 0, 8, 12, and 16 h after inoculation. Conidia were harvested and washed twice in 50 ml ice-cold sterile water, resuspended in 25 ml ice-cold YED (1% yeast extract, 1% glucose, w/v) plus 20 mM HEPES (adjusted to pH 8.0 with 100 mM Tris/HCl) and incubated at 26°C for 1 h on a rotary shaker at 100 rpm. Conidia were collected with centrifugation and resuspended in 2.5 ml (about 2 × 108 conidia ml−1) of ice-cold electroporation buffer (10 mM Tris/HCl, 200 mM sucrose, 1 mM lithium acetate, pH 7.5). Subsequently, 500 μl sterile glycerol was added into tube and mixed well. The resultant suspension in 50 μl aliquots was stored at −80°C and used as competent cells for electroporation transformation.
Electroporation transformation
One μg of pBFT DNA was linearized by HindIII, mixed with 50 μl competent cells in a total volume of 60 μl, and kept on ice for 15 min. The ice-cold suspension was transferred to 0.2 cm electroporation cuvette and electroporated at various settings with Gene Pulser apparatus (BioRad). After pulse, 1 ml ice-cold YED was immediately added into the electroporation cuvette, and the suspended cells were transferred to a 10 ml pre-cooled sterile tube and kept on ice for 15 min. The sample was then incubated at 26°C for 45 min on a rotary shaker at 100 rpm. The conidia were harvested by centrifugation and resuspended in 1 ml sterile deionized water. One hundred μl of suspension was spread on each Czapek’s plate containing 60 μg phosphinothricin ml−1 and incubated at 26°C. About 5 d later, the resistant colonies were transferred to a fresh Czapek’s plate with 60 μg phosphinothricin ml−1 for further analysis.
Southern blotting analysis
Genomic DNA of B. bassiana was extracted as previously described (Reader and Broda 1985). Southern blotting analysis was performed according to the method (Sambrook et al. 1989).
Mitotic stability analysis of transformants
Mitotic stability analysis was performed as previously described (Fang et al. 2006).
Results
Transformation frequencies were dependent on the germination stage of conidia
To determine the optimum germination time of conidia for transformation, the conidia morphology and nucleus number were observed at different germination stages. The conidia began to swell at 8 h after inoculation. At 12 h after incubation, the germ tube was formed at one end of the conidia and all germinated cells observed were found with single nucleus. After 16 h of incubation, the germinated cells had two or more nuclei (Fig. 2a).

Transformation frequencies were dependent on the germination stage of conidia. (a) The conidia of B. bassiana at 0, 8, 12, and 16 h after inoculation were stained with DAPI and viewed using a Leica D filter cube with peak transmission for excitation at 355–425 nm and suppression at above 470 nm. Bar = 10 μm. (b) The highest frequency was obtained with the field strength value of 5.5 kV cm−1 and the pulse duration of 4 ms. Capacitance and resistance was kept fixed at 25 μF and 400 ohms, respectively. Experiments were carried out in three independent experiments
To further confirm the optimum culture time for transformation, competent cells were harvested in the various germination stages of conidia and treated with 1 μg linear pBFT plasmid DNA. The highest transformation frequency (about 60 transformants μg−1 plasmid DNA) was achieved with conidia harvested at 12 h after inoculation (Fig. 2b). Based on the results, the germinated conidia cultivated for 12 h were employed to prepare competent cells for transformation.
Optimization of transformation conditions
To achieve high transformation efficiency, the field strength values (from 3.5 to 6.5 kV cm−1) and pulse duration (from 1 to 4 ms) were tested. The highest frequency (65 ± 7 transformants μg−1 plasmid DNA) was obtained with the field strength value of 5.5 kV cm−1 and the pulse duration of 4 ms (Table 2).
Effect of frozen storage on transformation frequency
The competent cells, containing glycerol stored at −80°C, were used daily to perform the transformation. No obvious difference in transformation frequency (about 60 transformants μg−1 plasmid DNA) during 1 month of storage was found (Fig. 3). However, 2 month storage led the transformation frequency dramatically decreasing (about 10 transformants μg−1 plasmid DNA).

Effect of frozen storage on transformation frequency. The competent cells stored at −80°C were used to perform the transformation at several-day intervals
Validation of transformants
Fluorescent observation showed that transformants selected by phosphinothricin resistance had GFP activity (Fig. 4a, b), indicating that BAR::EGFP fusion protein had both GFP and phosphinothricin resistant activity. Thus, the fusion gene bar::egfp could be used as selectable and visible marker. Seven transgenic lines were selected randomly for Southern blotting analysis. Strong hybridization bands appeared in transgenic lines and these seven transgenic lines contained a single copy insert of the transgene (Fig. 4c). To monitor mitotic stability of transformants, 30 randomly selected transformants were used. After five successive subcultures on nonselective media, most conidia and mycelia still displayed intensive green cytoplasmic fluorescence. Statistically, about 90% of the transformants were mitotically stable.

Validation of B. bassiana transformed with vector pBFT. Conidia and mycelia of transformants were imaged in the same microscopic field of light (a) and fluorescence (b). Bar = 10 μm. Southern blotting analysis of the transformants (c). About 5 μg genomic DNA of the wild-type strain Bb0062 (lane 1) and the transformants (lanes 2–8) was digested with DraI, fractionated on a 1% (w/v) agarose gel, transferred to a nylon membrane and hybridized with α-32P labeled bar. Molecular weight markers were indicated in the left margin.
Discussion
The results presented here showed that B. bassiana could be efficiently transformed by electroporation of germinated conidia using the fusion gene bar::egfp as selectable and visible marker. Compared to other transformation methods, the approach for B. bassiana transformation provided several advantages. First, it was significantly timesaving. It took about 18 h from initial inoculation to final plating on selective media, while other previous methods for B. bassiana needed at least 48 h of incubation (Daboussi et al. 1989; Fang et al. 2004; Pfeifer and Khachatourians 1992; Ying and Feng 2006). Second, it was simple and convenient. Like bacteria or yeast, after frozen at −80°C in 1 month, the germinated conidial cells were still fully competent for transformation, and the operation could be finished within 1.5 h. Third, it was highly efficient. In contrast with previously described methods such as polyethylene glycol (PEG)-mediated protoplast transformation (Daboussi et al. 1989; Pfeifer and Khachatourians 1992) and blastospore-based transformation (Jiang et al. 2007; Ying and Feng 2006), in which transformation frequencies were about 5–50 transformants μg−1 plasmid DNA, this method was able to obtain over 60 transformants μg−1 plasmid DNA. Finally, the fusion protein, BAR::EGFP, provided us a convenient maker for resistant selection and visible validation. Moreover, the engineered fungi could be monitored during various development or infection stages using this fusion gene.
References
Cho EM, Boucias D, Keyhani NO (2006a) EST analysis of cDNA libraries from the entomopathogenic fungus Beauveria (Cordyceps) bassiana. II. Fungal cells sporulating on chitin and producing oosporein. Microbiology 152:2855–2864
Cho EM, Liu L, Farmerie W et al (2006b) EST analysis of cDNA libraries from the entomopathogenic fungus Beauveria (Cordyceps) bassiana. I. Evidence for stage-specific gene expression in aerial conidia, in vitro blastospores and submerged conidia. Microbiology 152:2843–2854
Daboussi MJ, Djeballi A, Gerlinger C et al (1989) Transformation of seven species of filamentous fungi using the nitrate reductase gene of Aspergillus nidulans. Curr Genet 15:453–456
Fan Y, Fang W, Guo S et al (2007) Increased insect virulence in Beauveria bassiana strains overexpressing an engineered chitinase. Appl Environ Microbiol 73:295–302
Fang W, Zhang Y, Zheng X et al (2004) Agrobacterium tumefaciens-mediated transformation of Beauveria bassiana using an herbicide resistance gene as a selection marker. J Invertebr Pathol 85:18–24
Fang W, Leng B, Xiao Y et al (2005) Cloning of Beauveria bassiana chitinase gene Bbchit1 and its application to improve fungal strain virulence. Appl Environ Microbiol 71:363–370
Fang W, Pei Y, Bidochka MJ (2006) Transformation of Metarhizium anisopliae mediated by Agrobacterium tumefaciens. Can J Microbiol 52:623–626
Jiang Q, Ying SH, Feng MG (2007) Enhanced frequency of Beauveria bassiana blastospore transformation by restriction enzyme-mediated integration and electroporation. J Microbiol Methods 69:512–517
Koukaki M, Giannoutsou E, Karagouni A et al (2003) A novel improved method for Aspergillus nidulans transformation. J Microbiol Methods 55:687–695
Pfeifer TA, Khachatourians GG (1992) Beauveria bassiana protoplast regeneration and transformation using electroporation. Appl Microbiol Biotechnol 38:376–381
Reader U, Broda P (1985) Rapid preparation of DNA from filamentous fungi. Lett Appl Microbiol 1:17–20
Robinson M, Sharon A (1999) Transformation of the bioherbicide Colletotrichum gloeosporioides f. sp. aeschynomene by electroporation of germinated conidia. Curr Genet 36:98–104
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning. Cold Spring Harbor Laboratory Press, New York
Sánchez O, Aguirre J (1996) Efficient transformation of Aspergillus nidulans by electroporation of germinated conidia. Fungal Genet Newsl 43:48–51
Ying SH, Feng MG (2006) Novel blastospore-based transformation system for integration of phosphinothricin resistance and green fluorescence protein genes into Beauveria bassiana. Appl Microbiol Biotechnol 72:206–210
Acknowledgements
This work was supported by the grants from the Ministry of Science and Technology of China (2003CB114203 to Y. Pei) and the Natural Science Foundation of China (30471147 to Y. Zhang).
Author information
Authors and Affiliations
Corresponding author
Additional information
K. Jin and Y. Zhang contributed equally to this paper.
Rights and permissions
About this article
Cite this article
Jin, K., Zhang, Y., Luo, Z. et al. An improved method for Beauveria bassiana transformation using phosphinothricin acetlytransferase and green fluorescent protein fusion gene as a selectable and visible marker. Biotechnol Lett 30, 1379–1383 (2008). https://doi.org/10.1007/s10529-008-9713-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-008-9713-6