
Abstract
Heterologous expression of genes involved in the biosynthesis of various products is of increasing interest in biotechnology and in drug research and development. Microbial cells are most appropriate for this purpose. Availability of more microbial genomic sequences in recent years has greatly facilitated the elucidation of metabolic and regulatory networks and helped gain overproduction of desired metabolites or create novel production of commercially important compounds. Saccharomyces cerevisiae, as one of the most intensely studied eukaryotic model organisms with a rich density of knowledge detailing its genetics, biochemistry, physiology, and large-scale fermentation performance, can be capitalized upon to enable a substantial increase in the industrial application of this yeast. In this review, we describe recent efforts made to produce commercial secondary metabolites in Saccharomyces cerevisiae as pharmaceuticals. As natural products are increasingly becoming the center of attention of the pharmaceutical and nutraceutical industries, such as naringenin, coumarate, artemisinin, taxol, amorphadiene and vitamin C, the use of S. cerevisiae for their production is only expected to expand in the future, further allowing the biosynthesis of novel molecular structures with unique properties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Secondary metabolites of microbial, plant, animal and human origin play essential roles in the maintenance of human health and treatment of human disease (Newman et al. 2000). Many drugs being used in modern medicine, such as vinblastine (Madagascar periwinkle), digitalis (purple foxglove) and codeine (opium poppy), are derived from herbal remedies and used for diverse purposes in the treatment of cancer, heart diseases and pain. Natural products play a pivotal role as sources of drug lead compounds. Their chemical diversity and complexity provide structural scaffolds for small molecule drugs, serving as an inspiration for medicinal design. But on the other hand, chemical complexity of natural products also represents a major obstacle in the production of these pharmaceuticals on an industrial scale. Chemical synthesis of natural products is often difficult and expensive, and isolation from their natural resources is also typically low yielding.
The advent of molecular biology has offered a new platform for the research and development of natural products. Genetic manipulation of host strains such as E. coli or S. cerevisiae (Khosla and Keasling 2003) and heterologous expression allows for economical production of natural products produced in trace quantities in source organisms. Protein engineering and directed evolution can also be utilized to improve natural product yields. In addition, these techniques make it possible to manipulate biosynthetic pathways completely, and genes responsible for biosynthetic steps from a number of source organisms can be combined and modified to produce desired natural products or natural products with modified chemical structures that are rarely or not found in nature. These novel products with unique chemical properties may have improved efficacy or can be used for completely new purposes. In addition, emerging technologies such as metagenomics provide new approaches to accessing genes responsible for desired biosynthetic steps and potentialities for improving production of known natural products or isolation of useful novel compounds.
At present, microorganisms like bacteria, yeasts (Porro et al. 2005), especially Pichia (Gasser et al. 2006) and Kluyveromyces (Rodriguez et al. 2006), as well as cultured cells from higher organisms (such as mammalians, insects, or plants) represent the most frequently used hosts for the production of heterologous and homologous proteins. Microorganisms (prokaryotic as well as eukaryotic) are advantageous hosts because of high growth rates and easy of genetic manipulation.
Good examples of this are: (1) the use of a few selected microorganisms to produce a wide range of different enzymes (the Danish company Novozymes has expressed a large number of different enzymes in the filamentous fungus Aspergillus oryzae); (2) the use of the penicillin-producing fungus Penicillium chrysogenum by the Dutch company, DSM, for the production of adipoyl-7-aminodeacetoxycephalosporanic acid (adipoyl-7-ADCA) (Robin et al. 2001), a precursor for the production of semi-synthetic cephalosporins, and (3) the production of chemical 1,3-propanediol by the American company, Dupont, by a recombinant Escherichia coli, an organism that is already used for the production of many other chemicals, such as phenylalanine (Maury et al. 2005).
Among the microbial and eukaryotic host systems, yeasts combine the advantages of unicellular organisms (i.e., easy for genetic manipulation and growth) with the capability of protein processing typical for eukaryotic organisms (i.e., protein folding, assembly and post-translational modifications), together with the absence of endotoxins as well as oncogenic or viral DNA. The choice of the yeast host is of paramount importance for the success of the whole process. On this topic, some key reviews have been published (Gellissen and Hollenberg 1997; Sudbery 1996; Gellissen 2000; Dominguez et al. 1998; Cereghino and Cregg 2000; Giga-Hama and Kumagai 1999). Usually yeast hosts are divided in two main categories: (1) conventional and nonconventional or (2) Crabtree-positive and Crabtree-negative yeast hosts. In this respect, S. cerevisiae is the only conventional yeast and one of the few Crabtree positive (i.e. producing ethanol under aerobic conditions), together with Zygosaccharomyces rouxii and Z. bailii. As for other yeasts, S. cerevisiae is a reflection of the familiarity of molecular biologists with this yeast, combined with the deep knowledge about its genetics, biochemistry, physiology, and fermentation technologies. Furthermore, S. cerevisiae is recognized by the US Food and Drug Administration (FDA) as an organism generally regarded as safe (GRAS) (Chemler et al. 2006). Another important reason for the applicability of S. cerevisiae within the field of biotechnology is its susceptibility to genetic modifications by recombinant DNA technology, which has been even further facilitated by the availability of the complete genome sequence of S. cerevisiae, published in 1996 (Goffeau et al. 1996). The whole-genomes of S. cerevisiae have been public and annotated in the Saccharomyces Genome Database (http://www.yeastgenome.org/VL-yeast.html). A remarkable aspect of this wealth of information is that the number of genes that are expected to be involved in secondary metabolite production dramatically outnumbers the number of known secondary metabolites. So we can apply mutagenesis and recombinant DNA technology to increase the production of microbial products and generate new compounds. A genome-scale metabolic model serves such a purpose, providing a general description of the current state of knowledge regarding the genetics and biochemistry of metabolism in an organism (Murray 1991). Therefore, this wealth of information can be expected to make this species serve as a useful model for studying secondary metabolism.
In this perspective, we discuss ongoing research aimed toward the production of specific molecules that are used as pharmaceuticals in genetically engineered Saccharomyces cerevisiae.
Flavonoids and other phenylpropanoid-derived natural products
According to Metcalf (1987), between 50,000 and 100,000 plant secondary metabolite products may exist, and approximately 20% of the carbon fixed by photosynthesis is believed to be channeled into the phenylpropanoid pathway, generating the majority of the phenolic compounds found in nature, including flavonoids and stilbenoids (Ralston et al. 2005). Amongst these compounds, flavonoids are of exceptional interest and represent one of the most diverse and largest groups of plant natural products. Much of the increasing interest in flavonoids is owed to the beneficial properties they possess with respect to human health, as reviewed recently by Dixon (2004). The benefits of specific flavonoids and other phenylpropanoid-derived compounds to human health and their potential benefits for long-term health have only been recognized more recently. However, the productivity of flavonoids and stilbenoids is limited by the low concentrations and the low growth rates of plants. An obvious approach toward this end is to exploit the structural genes encoding enzymes of the general phenylpropanoid and particularly flavonoid and stilbenoid metabolism, whose coordinate expression leads to flavonoid or stilbenoid production as is listed in Table 1.
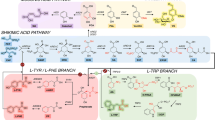
Ro and Douglas (2004) were the first to connect the two initial enzymes involved in the phenylpropanoid pathway, namely phenylalanine ammonia lyase (PAL) and cinnamate 4-hydroxylase (C4H) in S. cerevisiae together with a cytochrome P450 reductase (CPR). They evaluated metabolic flux into the phenylpropanoid pathway from phenylalanine to p-coumaric acid (Fig. 1). Later, Jiang et al. (2005) and Yan et al. (2007) reported biosynthesis of flavanones, the common precursors of the vast majority of flavonoids, in S. cerevisiae. Jiang et al. (2005) demonstrated the production of monohydroxylated naringenin and unhydroxylated pinocembrin at levels of 7 and 0.8 mg/l, respectively (Fig. 2). Yan et al. (2007) reported a four-step metabolic circuit that was constructed in S. cerevisiae with plant genes from heterologous origins: 4-coumarate:coenzyme A ligase (4CL) from Petroselinum crispum, chalcone synthases (CHS) from Medicago sativa and Petunia × hybrida and chalcone reductase (CHR) and chalcone isomerase (CHI) from M. sativa. Expression of the recombinant pathway in S. cerevisiae resulted in the accumulation of both 5-hydroxyflavanone and 5-deoxyflavanone. Other phenylpropanoid acid precursors, such as cinnamic acid and caffeic acid, could also be metabolized through the recombinant pathway, yielding corresponding 5-deoxyflavanone compounds.

Phenylalanine metabolism in yeast triple-expressing strains (transformed by genes coding for PAL, C4H, and CPR), showing reconstruction of the entry point into phenylpropanoid metabolism in this host. Endogenous yeast reactions involved in Phe (phenylalanine) catabolism are shaded in gray. The new pathway generated by introduction of PAL, C4H, and CPR genes is boxed in black, and endogenous pathways that metabolize cinnamate and p-coumarate are circled. ADH, alcohol dehydrogenase; DC, decarboxylase; PAD, phenylacrylic acid decarboxylase; TA, transaminase. Width of arrows is proportional to carbon flux, and potentially reversible reactions are indicated by double arrows. Question marks indicate potential but uncharacterized reactions or products

Proposed reactions catalyzed by S. cerevisiae AH22 coexpressing genes coding for PAL, 4CL, and CHS. PAL (TAL), phenylalanine (tyrosine) ammonialyase; 4CL, 4-coumarate: CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; Padp1, phenylacrylic acid decarboxylase. Overexpressed proteins are shown in bold. The biosynthesis of naringenin in plants is shown in the box
Recently, Ralston et al. (2005) reported the partial reconstruction of isoflavonoid biosynthesis in S. cerevisiae by using different types of CHI and an isoflavone synthase (IFS) from soybean (Glycine max).
Becker et al. (2003) succeeded in enhancing the production of stilbene resveratrol by making use of specifically transformed yeast strains. In this work, grape must was fermented with a yeast strain harboring two plasmids, one carrying a 4CL gene from poplar and the other carrying resveratrol synthase (RS) gene from grape. Transformed yeast strains were capable of converting p-coumaric acid from the medium into resveratrol glycosides. However, resveratrol glycosides were not synthesized at high concentrations (0.5–1.5 μg/l culture), where glycosides were synthesized instead of aglycons, and resveratrol was not released into the medium. Although this strategy needs to be optimized, it may provide a method for enhancing resveratrol content in fermented beverages (Pretorius and Bauer 2002; Vivier and Pretorius 2002).
Isoprenoids (terpenoid, terpene)
Isoprenoids with more than 40,000 described compounds (Withers and Keasling 2007) are the largest and most structurally diverse group of plant metabolites. Isoprenoids play various biological roles in plants. Depending on the number of isoprene units, isoprenoids can be classified into several groups, such as monoterpenes, sesquiterpenes and diterpenes (respectively two, three and four C5 units). It is among these secondary metabolites that many effective and promising pharmaceuticals, such as Taxol, vinblastine, artemisinin and prostratin, have been discovered and isolated.
Much work has focused on the introduction of exogenous isoprenoid metabolism into yeast listed in Table 2, such as the precursors to artemisinin (Ro et al. 2006) and Taxol (DeJong et al. 2006).
Geranyl pyrophosphate (GPP) is the universal precursor of monoterpenoids. In yeast it occurs exclusively as an intermediate of farnesyl pyrophosphate (FPP) synthesis. Oswald et al. (2007) overexpressed linalool synthase (LIS) from Clarkia breweri and geraniol synthase (GES) from Ocimum basilicum (sweet basil) in the wild-type (WT) and mutant strains to evaluate monoterpenol production levels in strains with normal or defective FPP biosynthesis for the objective of specific monoterpenol production–geraniol and linalool in S. cerevisiae (Fig. 3).

Biosynthesis pathway of geraniol and linalool from primary precursors dimethylallyl pyrophosphate (DMAPP), isopentenyl pyrophosphate (IPP) and farnesyl pyrophosphate (FPP). The indicated enzymes are: (1) farnesyl pyrophosphate synthase (FPPS); (2) geraniol synthase (GES); (3) linalool synthase (LIS); (4) geranyl pyrophosphate synthase (GPPS)
Takahashi et al. (2007) engineered yeast for sesquiterpene accumulation by introducing genetic modifications that enable the yeast to accumulate high levels of the key intermediate farnesyl pyrophosphate (FPP). Coexpression of terpene synthase genes diverted the enlarged FPP pool to greater than 80 mg of sesquiterpene/l; and finally the functional hydroxylation of a sesquiterpene scaffold by coexpression of a cognate cytochrome P450 hydroxylase and cytochrome P450 reductase, yielding 50 mg/l each of hydrocarbon and hydroxylated products (Fig. 4).

The mevalonate (MVA) pathway and those steps engineered in the current study for novel sesquiterpene production in yeast are boxed. The MVA pathway is localized to the cytoplasm in eukaryotic cells and supports the biosynthesis of numerous terpene macromolecules. In Saccharomyces cerevisiae, ergosterol is the dominant terpene derived from FPP, a highly regulated 15-carbon intermediate in the MVA pathway (Gardner and Hampton 1999). Various combinations of mutations in genes coding for a phosphatase (DPP1) and squalene synthase (ERG9), along with selection for aerobic uptake of exogenous ergosterol (sue) and engineering of a soluble, unregulated form of 3-hydroxy-3-methylglutaryl CoA reductase (HMGR) should yield yeast strains capable of accumulating excess levels of FPP. The potential to divert the excess FPP to novel sesquiterpene hydrocarbon biosynthesis was assessed by introducing key branch point enzymes (terpene synthases), and the potential to further modify these hydrocarbon skeletons was determined by cotransformation with a gene encoding for a cytochrome P450 hydroxylating enzyme (terpene hydroxylase)
Meanwhile, Asadollahi et al. (2007) chose S. cerevisiae as a microbial host for heterologous biosynthesis of three different plant sesquiterpenes: valencene, cubebol and patchoulol.
More interestingly, Ro et al. (2006) cloned a gene from A. annua that codes for a cytochrome P450. When this gene was expressed in yeast, amorpha-4, 11-diene was converted to artemisinic acid, which can be converted to artemisinin in a simple synthetic process (Fig. 5). Later, for high-level production of amorphadiene, a sesquiterpene precursor to the anti-malarial drug artemisinin was synthesized by the cyclization of FPP, Shiba et al. (2007) engineered the pyruvate dehydrogenase by passage in S. cerevisiae. Overproduction of acetaldehyde dehydrogenase and introduction of a Salmonella enterica acetyl-CoA synthetase variant increased the carbon flux into the mevalonate pathway, resulting in increased amorphadiene production.

Schematic representation of the engineered artemisinic acid biosynthetic pathway in S. cerevisiae strain EPY224 expressing CYP71AV1 and CPR in the box. Genes from the mevalonate pathway in S. cerevisiae that are directly upregulated are shown in blue (Chemler et al. 2006); those that are indirectly upregulated by upc2-1 expression are in purple; and the red line denotes repression of ERG9 in strain EPY224. The pathway intermediates IPP, DMAPP and GPP are defined as isopentenyl pyrophosphate, dimethyl allyl pyrophosphate and geranyl pyrophosphate, respectively. Green arrows indicate the biochemical pathway leading from farnesyl pyrophosphate (FPP) to artemisinic acid, which was introduced into S. cerevisiae from A. annua. The three oxidation steps converting amorphadiene to artemisinic acid by CYP71AV1 and CPR are shown
Recently, a partial taxol biosynthetic pathway was constructed in S. cerevisiae by expressing five sequential pathway steps leading from primary isoprenoid metabolism to intermediate taxadien-5α-acetoxy-10β-ol (DeJong et al. 2006). The Taxus cuspidate genes that the yeast host expressed included geranylgeranyl disphosphate synthase (GGPPS), taxadiene synthase (TS), cytochrome P450 taxadiene 5α-hydroxylase (TYH5a), taxadienol 5α-O-acetyl transferase (TAT) and taxoid 10β-hydroxylase (THY10b). The recombinant strain produced taxadiene at 1 mg/l while taxadien-5α-ol was produced in very small amounts (∼25 μg/l). These results suggest that the first two enzymes (GGPPS and TS) cooperated well with each other, and that the metabolic flux was reduced at the 5α-hydroxylation step, which is catalyzed by cytochrome P450 hydroxylase (DeJong et al. 2006). It is anticipated that overexpressing Taxus P450oxygenases with their corresponding P450 reductases in the yeast host would improve the overall production amounts (Jennewein et al. 2005).
Polyketides
Polyketides are a diverse group of natural products with significance in human and veterinary medicine. In their work, Mutka et al. (2006) describe the introduction into S. cerevisiae of pathways for the production of methylmalonyl-coenzyme A (CoA), a precursor for complex polyketides, by both propionyl-CoA-dependent and propionyl-CoA-independent routes. For propionyl-CoA-dependent methylmalonyl-CoA production, they expressed Salmonella typhimurium propionyl-CoA synthetase (PrpE) to catalyze the production of propionyl-CoA from propionate(included in the production medium) and endogenous ATP (Horswill and Escalante-Semerena 1999), and the propionyl-CoA carboxylase (PCC) pathway from Streptomyces coelicolor consisting of the transcarboxylase subunit, PccB, and the biotin carrier protein/biotin carboxylase subunit, AccA (Rodriguez and Gramajo 1999). Furthermore, they demonstrated the possibility of using methylmalonyl-CoA produced in the engineered yeast strains for the in vivo production of a polyketide product, triketide lactone (Fig. 6).

Pathways for the biosynthesis of propionyl-coenzyme A (CoA) and (2S)-methylmalonyl-CoA. Designations: PrpE: propionyl-CoA synthetase; PCC: propionyl-CoA carboxylase; PccB: encoding the transcarboxylase subunit of PCC complex; AccA: encoding the biotin carrier protein/biotin carboxylase subunit of PCC complex (Rodriguez and Gramajo 1999); MatB: malonyl/methylmalonyl-CoA ligase
Vitamin C
Most eukaryotic organisms produce l-ascorbic acid (l-AA or vitamin C), a water-soluble powerful antioxidant asscavenger of ROS (Levine 1986; Padh 1990, 1991) to prevent or at least alleviate deleterious effects caused by ROS. It is common knowledge that production of ROS is common in many types of cancer cells, and that vitamin C has a positive effect in reducing the occurrence of stomach, lung and colorectal cancer (Knekt et al. 1991; Valko et al. 2006). On the other hand, vitamin C can increase the generation of ROS under certain circumstances and could have detrimental effects at least under certain circumstances (Naidu 2003; O’Brien et al. 2003; Trommer et al. 2002). However, yeast cells naturally lack the ability to produce l-AA. Instead, erythro-ascorbic acid, a structurally related compound with chemical properties similar to those of l-AA, occurs at a low concentration in yeast cells (Huh et al. 1998). Its role in stress resistance has been established but to what extend it is important is still a question for scientific debate (Huh et al. 1998, 2001; Spickett et al. 2000). Animals and plants employ two different metabolic pathways to synthesize l-ascorbic acid (Wheeler et al. 1998; Banhegyi et al. 1997). Branduardi et al. (2007) reported for the first time the biosynthesis of vitamin C by recombinant S. cerevisiae cells starting from d-glucose utilizing a plant pathway (shown in Fig. 7). Accumulation of ascorbic acid was proven to be successful in two different strains, which were examined in parallel to elucidate the effect of a different genetic background.

Ascorbic acid biosynthetic pathway. Schematic representation of the pathway of l-AA production from d-glucose in plants. The following enzymes are involved: (A) hexokinase (2.7.1.1), (B) glucose-6-phosphate isomerase (5.3.1.9), (C) mannose-6-phosphate isomerase (5.3.1.8), (D) phosphomannomutase (5.4.2.8), (E) mannose-1-phosphate guanylyltransferase (2.7.7.22), (F) GDP-mannose-3,5-epimerase (5.1.3.18), (G) GDP-l-galactose phosphorylase (E.C.C not assigned), (H) l-galactose 1-phosphate phosphatase (3.1.3.25), (I) l-galactose dehydrogenase, (J) l-galactono-1,4-lactone dehydrogenase (1.3.2.3)
How many more secondary metabolites are produced by S. cerevisiae?
The present review shows that S. cerevisiae can produce a large number of secondary metabolites, including representatives of many of the principal classes of secondary metabolites found in nature. In addition, a variety of structures are often present from within a single class, a feature typical of secondary metabolism.
How many more secondary metabolites can be produced by S. cerevisiae? The progress in recent research suggests that many more new compounds would be produced in the next few years.
First of all, more biosynthetic pathways or secondary metabolism can be elucidated and more effective targets for genetic manipulation can be identified. Secondly, the spectrum of heterologous production is limited to a shortage of genes involved in secondary metabolic biosynthesis. There is a lack of information about enzymes involved in the biosynthesis of secondary metabolites, and mechanisms underlying the immense complex regulatory network of pathways have not been completely clarified. Improvements in gene synthesis technology will facilitate the construction of increasingly large sets of genes that comprise natural and artificial biosynthetic pathways, and less costly DNA sequencing technologies will drive the expansion of sequencing projects to identify biosynthetic genes for uncharacterized small molecules. Thirdly, a large number of transcription factors involved in secondary metabolic biosynthesis are better known, and the most recent biotechnological advances using genes encoding these factors have been reported. Further, considering the network of the biosynthetic pathway of plant secondary metabolites, the same metabolite can be a member of several different pathways and may also have regulatory effects on multiple biological processes. We can use combinatorial techniques to optimize the pathways, change the expression levels of an enzyme, engineer the specificity, alter post-translational regulation, or alter the metabolic flux through a particular biosynthetic pathway.
Conclusions and future directions
We herein introduce some recent developments of natural products in S. cerevisiae, and describe some emerging genetic manipulations to enrich the chemical diversity in nature. S. cerevisiae has enormous potential for the production of low and high molecular weight compounds as a heterologous host by expressing biosynthetic enzymes or even pathways.
The production of natural products in S. cerevisiae would probably provide cost-effective sources of drugs. Many top-selling drugs are natural products, accounting for approximately 40% of the top twenty drugs. It is anticipated that natural products will provide an increasing number of new drugs in the future, while engineering of biosynthetic enzymes will give access to a vast variety of “unnatural natural products” with improved properties or new biological activities. Developments in bioinformatics have speeded up the process of gene cloning and transformation. Besides, a number of powerful analytical techniques have been employed in metabolite analysis and analyses of cellular properties, which include gas chromatography and mass spectrometry(GC-MS), nuclear magnetic resonance (NMR), and high performance liquid chromatography fingerprint (HPLC fingerprint) (Nielsen 2001). Some of these different tools are reviewed below.
A key point in the study of heterologous production is the analysis at the cellular level to understand the metabolic function of biosynthetic enzymes in details, and this involves several different techniques (Nielsen 1998), including DNA chip and analytical techniques like GC-MS or liquid chromatography coupled with tandem mass spectrometry (LC-MS-MS). With enzyme assays, many different metabolites can be measured in complex matrices and with high sensitivity, as is illustrated in analysis of glycolytic intermediates of S. cerevisiae (Theobald et al. 1993, 1997; Vaseghi et al. 1999). The results may suggest the changes in flux distribution through different branches of the metabolic network. The methods and models used to calculate intracellular fluxes can now be directed toward determining how to manipulate cells to achieve the desired phenotype. After measuring the fluxes through the metabolic network, it is necessary to identify the pathways and enzymes that will most drastically improve the phenotype. Metabolic control analysis (MCA) provides a framework to help understand how flux control is distributed in a bioreaction network. Finding enzyme (gene) targets having the greatest influence on a product rate can be difficult because a rate-limiting step is often not found in biological networks. Instead, the limitations are spread over many enzymes in the network. The flux control coefficient (FCC) of an enzyme is defined as the relative effect of modulating the amount of an enzyme on the flux through the desired pathway. Genome-scale metabolic models for S. cerevisiae (Forster et al. 2003) have been developed in recent years and can be used in directed manipulation of the cellular network to predict changes that are required in the genotype of microorganisms in order to obtain efficient microbial strains (Patil et al. 2004).
These advances will improve our ability to control the shape and topology of secondary metabolites, allowing us to create new conformers that interact specifically with biological targets.
Future advances in directed biosynthesis would be driven by a mixture of technology and creativity. For example, the most interesting enzymes of secondary metabolism are expressed only during differentiation, and can be used in mixed chemical-enzymatic (transformation) production processes after overexpression in a microbial host, as is the case with 11β-hydroxylation. While metabolic engineering to increase the availability of FPP and diversion of this pool of intermediate to sesquiterpene hydrocarbon production is a critical and the first step in establishing any terpene production platform. Introduction of downstream biosynthetic modifications to the hydrocarbon scaffolds is of equal importance because of the structural complexity and biological activity afforded by these modifications (Takahashi et al. 2005). Many of these secondary modifications arise from the regio- and stereochemicalin sertion of hydroxyl groups into the hydrocarbon scaffolds, which then serve as sites for tertiary modifications like methylation, acetylation, aryl-group addition, and the formation of epoxides and lactones. Hence, efficient coupling of terpene hydroxylation reactions with hydrocarbon production is the first essential step for morecomplete recapitulation of the entire terpene biosynthetic repertoire.
References
Akashi T, Aoki T, Ayabe S (1999) Cloning and functional expression of a cytochrome P450 cDNA encoding 2-hydroxyisoflavanone synthase involved in biosynthesis of the isoflavonoid skeleton in licorice. Plant Physiol 121:821–828
Anzellotti D, Ibrahim RK (2004) Molecular characterization and functional expression of flavonol 6-hydroxylase. BMC Plant Biol 4:20
Asadollahi MA, Maury J, Moller K, Nielsen KF, Schalk M, Clark A, Nielsen J (2007) Production of plant sesquiterpenes in Saccharomyces cerevisiae: effect of ERG9 repression on sesquiterpene biosynthesis. Biotechnol Bioeng 99:666–677
Banhegyi G, Braun L, Csala M, Puskas F, Mandl J (1997) Ascorbate metabolism and its regulation in animals. Free Radic Biol Med 23:793–803
Becker JV, Armstrong GO, van der Merwe MJ, Lambrechts MG, Vivier MA, Pretorius IS (2003) Metabolic engineering of Saccharomyces cerevisiae for the synthesis of the wine-related antioxidant resveratrol. FEMS Yeast Res 4:79–85
Beekwilder J, Wolswinkel R, Jonker H, Hall R, de Vos CH, Bovy A (2006) Production of resveratrol in recombinant microorganisms. Appl Environ Microbiol 72:5670–5672
Branduardi P, Fossati T, Sauer M, Pagani R, Mattanovich D, Porro D (2007) Biosynthesis of vitamin C by yeast leads to increased stress resistance. PLoS ONE 2:e1092
Cereghino JL, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66
Chemler JA, Yan Y, Koffas MA (2006) Biosynthesis of isoprenoids, polyunsaturated fatty acids and flavonoids in Saccharomyces cerevisiae. Microb Cell Fact 5:20
Dejong JM, Liu Y, Bollon AP, Long RM, Jennewein S, Williams D, Croteau RB (2006) Genetic engineering of taxol biosynthetic genes in Saccharomyces cerevisiae. Biotechnol Bioeng 93:212–224
Dixon RA (2004) Phytoestrogens. Annu Rev Plant Physiol Plant Mol Biol 55:225–261
Dominguez A, Ferminan E, Sanchez M, Gonzalez FJ, Perez-Campo FM, Garcia S, Herrero AB, San Vicente A, Cabello J, Prado M, Iglesias FJ, Choupina A, Burguillo FJ, Fernandez-Lago L, Lopez MC (1998) Non-conventional yeasts as hosts for heterologous protein production. Int Microbiol 1:131–142
Forster J, Famili I, Fu P, Palsson BO, Nielsen J (2003) Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res 13:244–253
Gardner RG, Hampton RY (1999) A highly conserved signal controls degradation of 3-hydroxy-3-methylglutaryl-coenzyme a (HMG-CoA) reductase in eukaryotes. J Biol Chem 274:31671–31678
Gasser B, Maurer M, Gach J, Kunert R, Mattanovich D (2006) Engineering of Pichia pastoris for improved production of antibody fragments. Biotechnol Bioeng 94:353–361
Gellissen G (2000) Heterologous protein production in methylotrophic yeasts. Appl Microbiol Biotechnol 54:741–750
Gellissen G, Hollenberg CP (1997) Application of yeasts in gene expression studies: a comparison of Saccharomyces cerevisiae, Hansenula polymorpha and Kluyveromyces lactis—a review. Gene 190:87–97
Giga-Hama Y, Kumagai H (1999) Expression system for foreign genes using the fission yeast Schizosaccharomyces pombe. Biotechnol Appl Biochem 30(Pt 3):235–244
Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG (1996) Life with 6000 genes. Science 274:546, 563–547
Hezari M, Lewis NG, Croteau R (1995) Purification and characterization of taxa-4(5),11(12)-diene synthase from Pacific yew (Taxus brevifolia) that catalyzes the first committed step of taxol biosynthesis. Arch Biochem Biophys 322:437–444
Horswill AR, Escalante-Semerena JC (1999) The prpE gene of Salmonella typhimurium LT2 encodes propionyl-CoA synthetase. Microbiology 145(Pt 6):1381–1388
Huh WK, Lee BH, Kim ST, Kim YR, Rhie GE, Baek YW, Hwang CS, Lee JS, Kang SO (1998) d-Erythroascorbic acid is an important antioxidant molecule in Saccharomyces cerevisiae. Mol Microbiol 30:895–903
Huh WK, Kim ST, Kim H, Jeong G, Kang SO (2001) Deficiency of d-erythroascorbic acid attenuates hyphal growth and virulence of Candida albicans. Infect Immun 69:3939–3946
Jennewein S, Park H, DeJong JM, Long RM, Bollon AP, Croteau RB (2005) Coexpression in yeast of Taxus cytochrome P450 reductase with cytochrome P450 oxygenases involved in Taxol biosynthesis. Biotechnol Bioeng 89:588–598
Jiang H, Wood KV, Morgan JA (2005) Metabolic engineering of the phenylpropanoid pathway in Saccharomyces cerevisiae. Appl Environ Microbiol 71:2962–2969
Khosla C, Keasling JD (2003) Metabolic engineering for drug discovery and development. Nat Rev Drug Discov 2:1019–1025
Kim DH, Kim BG, Lee HJ, Lim Y, Hur HG, Ahn JH (2005) Enhancement of isoflavone synthase activity by co-expression of P450 reductase from rice. Biotechnol Lett 27:1291–1294
Knekt P, Jarvinen R, Seppanen R, Rissanen A, Aromaa A, Heinonen OP, Albanes D, Heinonen M, Pukkala E, Teppo L (1991) Dietary antioxidants and the risk of lung cancer. Am J Epidemiol 134:471–479
Levine M (1986) New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med 314:892–902
Maury J, Asadollahi MA, Moller K, Clark A, Nielsen J (2005) Microbial isoprenoid production: an example of green chemistry through metabolic engineering. Adv Biochem Eng Biotechnol 100:19–51
Mercke P, Crock J, Croteau R, Brodelius PE (1999) Cloning, expression, and characterization of epi-cedrol synthase, a sesquiterpene cyclase from Artemisia annua L. Arch Biochem Biophys 369:213–222
Mercke P, Bengtsson M, Bouwmeester HJ, Posthumus MA, Brodelius PE (2000) Molecular cloning, expression, and characterization of amorpha-4,11-diene synthase, a key enzyme of artemisinin biosynthesis in Artemisia annua L. Arch Biochem Biophys 381:173–180
Metcalf RL (1987) Plant volatiles as insect attractants. CRC Crit Rev Plant Sci 5:251–301
Murray RDH (1991) Progress in the chemistry of organic natural products. Springer Wien, New York
Mutka SC, Bondi SM, Carney JR, Da Silva NA, Kealey JT (2006) Metabolic pathway engineering for complex polyketide biosynthesis in Saccharomyces cerevisiae. FEMS Yeast Res 6:40–47
Naidu KA (2003) Vitamin C in human health and disease is still a mystery? An overview. Nutr J 2:7
Newman DJ, Cragg GM, Snader KM (2000) The influence of natural products upon drug discovery (Antiquity to late 1999). Nat Prod Rep 17:215–234
Nielsen J (1998) Metabolic engineering: techniques for analysis of targets for genetic manipulations. Biotechnol Bioeng 58:125–132
Nielsen J (2001) Metabolic engineering. Appl Microbiol Biotechnol 55:263–283
O’Brien TJ, Ceryak S, Patierno SR (2003) Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res 533:3–36
Oswald M, Fischer M, Dirninger N, Karst F (2007) Monoterpenoid biosynthesis in Saccharomyces cerevisiae. FEMS Yeast Res 7:413–421
Padh H (1990) Cellular functions of ascorbic acid. Biochem Cell Biol 68:1166–1173
Padh H (1991) Vitamin C: newer insights into its biochemical functions. Nutr Rev 49:65–70
Patil KR, Akesson M, Nielsen J (2004) Use of genome-scale microbial models for metabolic engineering. Curr Opin Biotechnol 15:64–69
Porro D, Sauer M, Branduardi P, Mattanovich D (2005) Recombinant protein production in yeasts. Mol Biotechnol 31:245–259
Pretorius IS, Bauer FF (2002) Meeting the consumer challenge through genetically customized wine-yeast strains. Trends Biotechnol 20:426–432
Ralston L, Subramanian S, Matsuno M, Yu O (2005) Partial reconstruction of flavonoid and isoflavonoid biosynthesis in yeast using soybean type I and type II chalcone isomerases. Plant Physiol 137:1375–1388
Ro DK, Douglas CJ (2004) Reconstitution of the entry point of plant phenylpropanoid metabolism in yeast (Saccharomyces cerevisiae): implications for control of metabolic flux into the phenylpropanoid pathway. J Biol Chem 279:2600–2607
Ro DK, Paradise EM, Ouellet M et al (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943
Robin J, Jakobsen M, Beyer M, Noorman H, Nielsen J (2001) Physiological characterisation of Penicillium chrysogenum strains expressing the expandase gene from Streptomyces clavuligerus during batch cultivations. Growth and adipoyl-7-aminodeacetoxycephalosporanic acid production. Appl Microbiol Biotechnol 57:357–362
Rodriguez E, Gramajo H (1999) Genetic and biochemical characterization of the alpha and beta components of a propionyl-CoA carboxylase complex of Streptomyces coelicolor A3(2). Microbiology 145(Pt 11):3109–3119
Rodriguez AP, Leiro RF, Trillo MC, Cerdan ME, Siso MI, Becerra M (2006) Secretion and properties of a hybrid Kluyveromyces lactis-Aspergillus niger beta-galactosidase. Microb Cell Fact 5:41
Schoenbohm C, Martens S, Eder C, Forkmann G, Weisshaar B (2000) Identification of the Arabidopsis thaliana flavonoid 3′-hydroxylase gene and functional expression of the encoded P450 enzyme. Biol Chem 381:749–753
Shiba Y, Paradise EM, Kirby J, Ro DK, Keasling JD (2007) Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae for high-level production of isoprenoids. Metab Eng 9:160–168
Spickett CM, Smirnoff N, Pitt AR (2000) The biosynthesis of erythroascorbate in Saccharomyces cerevisiae and its role as an antioxidant. Free Radic Biol Med 28:183–192
Sudbery PE (1996) The expression of recombinant proteins in yeasts. Curr Opin Biotechnol 7:517–524
Takahashi S, Zhao Y, O’Maille PE et al (2005) Kinetic and molecular analysis of 5-epiaristolochene 1,3-dihydroxylase, a cytochrome P450 enzyme catalyzing successive hydroxylations of sesquiterpenes. J Biol Chem 280:3686–3696
Takahashi S, Yeo Y, Greenhagen BT et al (2007) Metabolic engineering of sesquiterpene metabolism in yeast. Biotechnol Bioeng 97:170–181
Theobald U, Mailinger W, Reuss M, Rizzi M (1993) In vivo analysis of glucose-induced fast changes in yeast adenine nucleotide pool applying a rapid sampling technique. Anal Biochem 214:31–37
Theobald U, Mailinger W, Baltes M, Rizzi M, Reuss M (1997) In vivo analysis of metabolic dynamics in Saccharomyces cerevisiae. I. Experimental observations. Biotechnol Bioeng 55:305–316
Tian L, Dixon RA (2006) Engineering isoflavone metabolism with an artificial bifunctional enzyme. Planta 224:496–507
Trommer H, Bottcher R, Poppl A, Hoentsch J, Wartewig S, Neubert RH (2002) Role of ascorbic acid in stratum corneum lipid models exposed to UV irradiation. Pharm Res 19:982–990
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160:1–40
Vannelli T, Wei Qi W, Sweigard J, Gatenby AA, Sariaslani FS (2007) Production of p-hydroxycinnamic acid from glucose in Saccharomyces cerevisiae and Escherichia coli by expression of heterologous genes from plants and fungi. Metab Eng 9:142–151
Vaseghi S, Baumeister A, Rizzi M, Reuss M (1999) In vivo dynamics of the pentose phosphate pathway in Saccharomyces cerevisiae. Metab Eng 1:128–140
Vivier MA, Pretorius IS (2002) Genetically tailored grapevines for the wine industry. Trends Biotechnol 20:472–478
Wheeler GL, Jones MA, Smirnoff N (1998) The biosynthetic pathway of vitamin C in higher plants. Nature 393:365–369
Withers ST, Keasling JD (2007) Biosynthesis and engineering of isoprenoid small molecules. Appl Microbiol Biotechnol 73:980–990
Yan Y, Kohli A, Koffas MA (2005) Biosynthesis of natural flavanones in Saccharomyces cerevisiae. Appl Environ Microbiol 71:5610–5613
Yan Y, Huang L, Koffas MA (2007) Biosynthesis of 5-deoxyflavanones in microorganisms. Biotechnol J 2:1250–1262
Zhang Y, Li SZ, Li J, Pan X, Cahoon RE, Jaworski JG, Wang X, Jez JM, Chen F, Yu O (2006) Using unnatural protein fusions to engineer resveratrol biosynthesis in yeast and Mammalian cells. J Am Chem Soc 128:13030–13031
Acknowledgements
This research was funded by Shanghai Science and Technology Committee (No. 054319936) and the National Natural Science Foundation of China (No. 20702062).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Huang, B., Guo, J., Yi, B. et al. Heterologous production of secondary metabolites as pharmaceuticals in Saccharomyces cerevisiae . Biotechnol Lett 30, 1121–1137 (2008). https://doi.org/10.1007/s10529-008-9663-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-008-9663-z