
We obtained evidence from a large study in Dutch twins (N=8387) and siblings (N=2295) that variation in loneliness has a genetic component. The heritability estimate for loneliness, which was assessed as an ordinal trait, was 40% and did not differ between males and females. There were 682 sibling pairs with genotypic (around 400 microsatellite markers) data. We combined phenotypic and genotypic data to carry out a genome scan to localize QTLs for loneliness. One region on chromosome 12q23.3–24.3, showed near suggestive linkage. Genetic association tests within this region revealed significant association (p-value 0.009) with one of the alleles of marker D12S79 and with one of the alleles of neighbouring marker D12S395 (p-value 0.043). We review evidence for linkage in this region for psychiatric disorders and discuss our findings within this context.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Loneliness has been described by Weiss (1973) as “a gnawing ... chronic disease without redeeming features” which may occur when one’s intimate and social needs are not adequately met (Baumeister and Leary, 1995). The core experience of loneliness consists of social isolation and the absence of both relational and collective connectedness (Hawkley et al., 2005; Russell et al., 1980). There is now substantial evidence that loneliness is at the heart of a constellation of socio-emotional states, which include self-esteem, mood, anxiety, anger, optimism, fear of negative evaluation, shyness, social skills, social support, dysphoria, and sociability (see reviews by Duck et al., 1994; Ernst and Cacioppo, 1999; Peplau and Perlman, 1982; Berscheid and Reis, 1998; Rook, 1988; Shaver and Brennan, 1991).
Recently, attention has been paid to how genetic and environmental factors influence the development of individual differences in loneliness. McGuire and Clifford (2000) examined the heritability of loneliness in children and found significant genetic (h 2=55% and 48%, respectively) contributions to individual differences in loneliness. To examine the genetic contribution to variation in loneliness in adults, Boomsma et al. (2005) conducted a study based on data of 8387 adult twins from the Netherlands Twin Register. It was found that individual differences in loneliness demonstrated considerable temporal stability and heritability. The estimate of heritability was 48%, and all environmental influences were unique to each individual. The genetic contributions to adult loneliness were similar in males and females and in a younger and an older cohort. Finally, no qualitative sex differences in heritability were found, indicating that the same genes influence loneliness in both sexes.
The heritability of loneliness is comparable to that of other complex traits which have been the subject of genome scans to identify susceptibility loci. As is the case for other complex traits, heritability may be caused by polymorphisms in several genes. Because a large number of genetic and environmental factors may contribute to the liability of loneliness, isolating the genetic component is a daunting undertaking (Stenger et al., 2005). We collected genotypic marker data and phenotypic information in 682 sibling pairs and their parents and aim, through linkage and association analysis, to elucidate candidate regions which may harbor genes influencing variation in loneliness. Loneliness was analyzed as an ordinal phenotype, using a threshold model which assumes a latent normally distributed liability (Falconer, 1989; Lynch and Walsh, 1998). The ordinal phenotype and the continuous liability are linked through thresholds and the liability was treated as an unobservable quantitative trait (Yi et al. 2004).
Methods
Participants and Phenotypic Information
In 1991 the Netherlands Twin Register (NTR) started a longitudinal survey study of health and lifestyle in adolescent and adult twins and their family members (Boomsma et al., 2002). Surveys were mailed out in 1991, 1993, 1995, 1997, 2000 and 2002/3. Twins were asked to participate in all waves, siblings were included since 1995. Five surveys contained items on loneliness from the YASR (Achenbach, 1990), which could be answered on a 3-point scale. Based on factor analyses, we selected 2 items (item 12: I feel lonely, and item 33: Nobody loves me). Information on loneliness was quantified by averaging over items and over years of observation (Boomsma et al., 2005). There were phenotypic data for 8387 twins, and for 2295 full siblings.
DNA Collection and Genotype Data
Genotype data came from 2 studies: a cardiovascular study of unselected DZ twin pairs (Heijmans et al., 2005) and a study of DZ twin and sibling pairs (Boomsma et al., 2000) who were selected for anxious depression. In the cardiovascular study, DNA was collected from blood samples. For 234 individuals, there were phenotypic data on loneliness. Genotyping in these samples was carried out by the Dept of Molecular Epidemiology (Leiden). A 419 marker genome scan (8.34 cM spacing) was done using a combination of in-house markers (ALFexpress automated sequencer (Amersham Pharmacia Biotech)) as described by Beekman et al. (2003), markers from the Weber screening set 8 (ALFexpress) and from the Human Linkage Set v2.5 MD10 and HD5 (ABI Prism DNA Analyzer 3700 (Applied Biosystems)).
DNA from buccal swabs was collected as part of the NETSAD project (Boomsma et al., 2000) in sibling pairs selected for anxious depression, with additional, non-extreme siblings in the family included in the DNA collection. For 523 siblings (50 of which were also genotyped as part of the cardiovascular study) phenotypic loneliness data were available. Genotyping in the NETSAD samples was performed by the Mammalian genotyping service (Marshfield, USA). A 379 autosomal marker genome scan (9.44 cM spacing) was done using microsatellite screening set 10 (Yuan et al. 1997) with few alternative markers. Excessive recombination rates were observed for five markers indicating potential problems. Those markers were not included in the analysis (D1S468; D1S1627; D11S1985; D11S2006 and D20S159-UT1307). Five other markers are currently inconsistently mapped and were also excluded. There were 100 markers typed in both scans. Mendelian errors were detected using PEDSTATS and unlikely double recombinants using MERLIN; both types of error were removed using PEDWIPE (Abecasis et al., 2002). Pedigree relationships were checked with GRR (Abecasis et al., 2001). Marker location was taken from an integrated genetic map with interpolated genetic map positions (http://www.2.qimr.edu.au/davidD/). The position is in Decode cM (Kong et al., 2002), estimated via locally weighted linear regression from the Build 34.3 (and 35.1) physical map positions and published Decode and Marshfield genetic map positions. Parents were typed for between 185 and 375 markers (mean=352, SD=33). For offspring, the number of typed markers ranged from 184 to 678, with an average of 378 (SD=78). Genotypic data as well as data on loneliness were available for 707 offspring, from 277 families (for 102 families both parents were genotyped and for 40 families one parent was typed), forming 682 sib pairs.
QTL Analysis
The phenotypic data were analyzed with a threshold model, with 3 thresholds dividing the liability to loneliness into 4 categories, and different thresholds for males and females (Boomsma et al., 2005). Because the genotyped pairs are not a random sample, thresholds were fixed at the estimates from the model in which data from all twins and siblings were analyzed. Variation in liability to loneliness was decomposed into variation due to a QTL ( \(\upsigma^2_{q}\)), additive polygenic influences ( \(\upsigma^2_{a}\)), and non-shared environmental influences ( \(\upsigma^2_{e}\)), using structural equation modelling as implemented in Mx (Neale et al., 2003). Estimates of the variance component associated with a putative QTL were obtained by using a \(\hat{\uppi}\) approach, in which the covariance due to the QTL for a sib-pair is modelled as a function of the estimated proportion of alleles shared identical by descent. The variance–covariance matrix for pair j,k of the i-th family (Ω ijk ) is given by:
\(\upsigma^2_{a}\), \(\upsigma^2_{q}\) and \(\upsigma^2_{e}\) denote the background and the QTL additive genetic and environmental variances. For DZ twin and sib pairs \(\uprho=1/2\) (twice the kinship coefficient). For DZ and sibling pairs \(\hat{\uppi}\) depends on the IBD status of the pair and is obtained as: \(\hat{\uppi}=0.5p_{\rm (IBD=1)}+p_{\rm (IBD=2)}\) where p (IBD=1) denotes the probability that the pair is IBD=1 and p (IBD=2) denotes the probability that the pair is IBD=2. Estimates for IBD probabilities were obtained from Merlin (Abecasis et al. 2002). Significance of variation of the QTL was evaluated by the likelihood ratio test, from which the LOD score was calculated (Sham, 1998) by dividing the test statistic \(\upchi^2\) by 2 ln 10 ( \(\sim4.6\)).
Permutation Tests
To obtain empirical estimates of genome-wide significance levels, 1000 permutations of the dataset were performed, keeping both the family structure and the IBD structure intact. These permutations account for uneven marker spacing and informativeness (Churchill and Doerge, 1994). Permuted datasets were obtained as follows: Each row in the observed dataset represents one family and contains phenotypes and IBD probabilities across the whole genome for all pairs within that family. This file is split into a phenotype file and an IBD file. Families are labeled with unique numbers one through n. The phenotypic data are then shuffled by taking a random permutation of the indices 1, ..., n and matching the ith phenotypic trait value to the family with index given by the ith element of permuted indices. This permuted vector of traits is matched with the original (non-permuted) genotypic information for all families. One thousand permuted datasets were generated; each permuted dataset was then analyzed. The threshold for suggestive linkage was calculated by recording the maximum LOD-score for each chromosome in 1000 runs, and determining what LOD-score occurs a 1000 times out of 22,000. This represents the average maximum peak size expected once per genome scan (Nyholt et al., 2005). The threshold for suggestive linkage for the ordinal trait loneliness was 1.58. The significant linkage threshold obtained by determining the maximum LOD-score for each scan, and was then defined as the LOD-score occurring in 50 of the 1000 permutations corresponding to a probability of 0.05 in a genome scan (Churchill and Doerge, 1994; Lander and Kruglyak, 1995). The threshold for significant linkage was 2.87.
Genetic Association Testing
Genetic association tests were conducted with the extended TDT test proposed by Monks and Kaplan (2000) and implemented in the program QTDT (Abecasis et al., 2000). This test uses a weighing scheme that provides a conservative test of association in families with multiple offspring, and uses parental genotypes if available. Because the program does not handle ordinal data with >2 categories, the ordinal variable was dichotomized to obtain an affection status by coding all individual with ordinal category 0 (i.e. always replied “no” to the 2 loneliness items) as unaffected, and all individuals with ordinal categories >0 as affected.
Results
The previous study of loneliness included data from twins only. We repeated the heritability analyses of loneliness for twins (N=8387) and their siblings. A maximum of two brothers and two sisters was added to the data set (data from additional brothers (N=33) and sisters (N=66) were not used). This added a total of 1019 brothers and 1276 sisters to the analyses. The heritability was estimated at 40% (95% CI=0.35–0.44). The prevalence for males and females in the sample that is available for linkage and in the total samples is given in Table I.
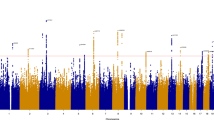
Figure 1 shows the results of the whole genome scan. One region, 12q23.3–24.3, showed a LOD-score (1.38) just below the empirical threshold for suggestive linkage. The drop 1 LOD-score borders are defined by markers D12S78 and D12S2078, which are 37 cM apart (see Figure 2).

Multipoint variance-component linkage of the 22 autosomes in 682 sib-pairs for loneliness. The X-axis plots genetic distance (in cM Haldane), and the Y-axis represents the LOD-score.

Best evidence of linkage to loneliness on chromosome 12. The X-axis plots genetic distance (in cM Haldane), and the Y-axis the LOD-score. Markers are arrayed in map order along the top.
We performed genetic association tests for loneliness’ affection status with microsatellite markers within the 12q23.3–24.3 region. These markers included D12S1300 and D12S2078, and all markers in between, as indicated in Figure 2 (N=9 markers; all markers were in Hardy–Weinberg equilibrium). We obtained a significant association between the marker allele with repeat length 169 of marker D12S79 (p-value 0.0094), and suggestive association within the same marker and allele with repeat length 157 (p-value 0.0656). For neighbouring marker D12S395 association was seen with the 235 repeat allele (p-value 0.0432), and suggestive association within the same marker and allele with repeat length 227 (p-value 0.0662). For the marker allele with repeat length 169 of marker D12S79 we observed that 54% of carriers of at least one allele was affected (i.e. scored non-zero on the loneliness trait), whereas 38% of subjects without the 169 allele was affected.
Discussion
A genome-wide scan for loneliness was conducted by treating loneliness as an ordinal phenotype, using a threshold model assuming a latent continuous liability. Considering the moderate heritability of loneliness and the general reduced power of ordinal data versus continuous data the odds of finding significant linkage seemed rather low at the outset. However, the Lander and Kruglyak (1995) guidelines for suggestive (2.2) and significant (3.6) linkage apply to quantitative traits, and may be too stringent for ordinal traits. Permutation testing confirmed that the thresholds for suggestive and significant linkage were considerably lower, i.e. 1.58 and 2.87. We obtained one near suggestive result from a genome-wide linkage scan for loneliness for a region on chromosome 12q23–24. Genetic association tests with markers within this area showed significant association to marker D12S79 and to neighbouring marker D12S395, offering support for the linkage result.
This region on chromosome 12 has been implicated before in studies of psychiatric disorders. Table II summarizes the evidence for linkage in this region from studies of psychiatric disease, including bipolar disorder, major depression, schizophrenia and indices of anxiety, neuroticism and alcohol abuse (see also Craddock et al., 2005; Hamet and Tremblay, 2005). This region on chromosome 12q was first implied at the chromosomes 12 and 16 workshop (Detera-Wadleigh, 1999) as a region with compelling evidence for a bipolar disorder susceptibility locus. This suggestion was further strengthened by the finding that Darier’s disease, which maps to this region, co-segregates with bipolar disorder. However, there is strong evidence against the Darier causing mutation itself being the susceptibility risk factor for bipolar disease (Jones et al., 2002). Neither was any evidence found for the DUSP6 gene on chromosome 12q23 (Toyota et al., 2000). Shink et al., (2005a, b) investigated the 12q24.31 region by saturating a 7.7 Mb genomic region with 20 microsatellite markers and obtained linkage support for bipolar disorder in this area. They next analyzed 32 genes for polymorphisms in coding sequences and intron/exon junctions. No strong support for any of the genes was found. A positive result was obtained recently with a haplotype in the gene encoding a transcription factor (regulatory factor X4: RFX4) on 12q23 by Glaser et al. (2005a). Their haplotype-bipolar association was supported by an association with microsatellite D12S2072. This marker, which was not included in our genome scan, is located between D12S78 and D12S2070/79. The same group (Glaser et al., 2005b) also observed association in this region with Cux2, which is a potential regulator of neural cell adhesion molecule expression, and with hypothetical protein FLJ32356; but not with PAH (Green et al., 2003), which we also investigated and for which we found no association.
The region on 12q23–q24 may contain a general vulnerability locus for psychiatric disorders. This possibility is reinforced by the finding of linkage for Neuroticism in this region. The 12q23–q24 region was, however, not reported in meta-analyses of bipolar disorder (Segurado et al., 2003) or schizophrenia (Lewis et al., 2003). Since the publication of the meta-analyses several further genome scans were published (see Table II) with at least 2 genome scans reporting genome wide significance (Ewald et al., 2002; Shink et al., 2005a, b) within the 12q23–24 region.
Understanding the origins of variation in loneliness may be important for clinical as well as scientific purposes. Loneliness is associated with psychiatric and behavioral problems, including bipolar disorder, depression (Eisemann, 1984; 1985; Segrin, 1998), alcoholism (Akerlind and Hornquist, 1992; Bell, 1956), impaired sleep (Cacioppo et al., 2002a, b), and suicide (Goldsmith et al., 2002; Wenz, 1977). Patients with these disorders are socially isolated and have difficulties in maintaining friendships and relations. Loneliness may be a consequence as well as a predictor of psychiatric problems. Heikkinen and Kauppinen (2004) reported that loneliness predicted changes in depressive symptoms in a 10-year study of elderly Finns and Cacioppo et al. (in press) found that loneliness predicted subsequent changes in depressive symptoms and that there was evidence for reciprocal influences over time. The results of the current study suggest that alleles in LD with an allele at microsatellite marker D12S79 and at neighbouring marker D12S395 may contribute to individual differences in loneliness. Although additional research is required to determine their common phenotypic expression, it is possible that genes in this region may be involved in both affective and social regulation and dysregulation.
References
Abecasis G. R., Cookson W. O., Cardon L. R. (2000). Pedigree tests of transmission disequilibrium. Eur. J. Hum. Genet. 8:545–551
Abecasis G. R., Cherny S. S., Cookson W. O., Cardon L. R. (2001). GRR: graphical representation of relationship errors. Bioinformatics 17:742–743
Abecasis G. R., Cherny S. S., Cookson W. O., Cardon L. R. (2002). Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat.Genet 30:97–101
Abkevich V., Camp N. J., Hensel C. H., Neff C. D., Russell D. L., Hughes D. C., Plenk A. M., Lowry M. R., Richards R. L., Carter C., Frech G. C., Stone S., Rowe K., Chau C. A., Cortado K., Hunt A., Luce K., O’Neil G., Poarch J., Potter J., Poulsen G. H., Saxton H., Bernat-Sestak M., Thompson V., Gutin A., Skolnick M. H., Shattuck D., Cannon-Albright L. (2003). Predisposition locus for major depression at chromosome 12q22–12q23.2. Am. J. Hum. Genet. 73:1271–1281
Achenbach T. M. (1990). The Young Adult Self Report. University of Vermont, Department of Psychiatry, Burlington, VT
Akerlind I., Hornquist J. O. (1992). Loneliness and alcohol abuse: A review of evidences and an interplay. Soc. Sci. Med. 34:405–414
Bailer U., Leisch F., Meszaros K., Lenzinger E., Willinger U., Strobl R., Heiden A., Gebhardt C., Doge E., Fuchs K., Sieghart W., Kasper S., Hornik K., Aschauer H. N. (2002). Genome scan for susceptibility loci for schizophrenia and bipolar disorder. Biol. Psychiatry. 52:40–52
Baumeister R. F., Leary M. R. (1995). The need to belong: Desire for interpersonal attachment as a fundamental human motivation. Psychol. Bull. 117:497–529
Beekman M., Heijmans B. T., Martin N. G., Whitfield J. B., Pedersen N. L., DeFaire U., Snieder H., Lakenberg N., Suchiman H. E., de Knijff P., Frants R. R., van Ommen G. J., Kluft C., Vogler G. P., Boomsma D. I., Slagboom P. E. (2003). Evidence for a QTL on chromosome 19 influencing LDL cholesterol levels in the general population. Eur. J. Hum. Genet. 11:845–850
Bell R.G. (1956). Alcoholism and loneliness. J. Soc. Therapy 2:171–181
Berscheid E., Reis H. T. (1998). Attraction and close relationships. In: Gilbert D. T., Fiske S. T. et al. (eds), The Handbook of Social Psychology: Vol. 2 4th ed., New York, McGraw Hill, pp. 193–281
Boomsma D. I., Beem A. L., van den B. M., Dolan C. V., Koopmans J. R., Vink J. M., de Geus E. J., and Slagboom P. E. (2000). Netherlands twin family study of anxious depression (NETSAD). Twin Res. 3:323–334
Boomsma D. I., Vink J. M., van Beijsterveldt T. C., de Geus E. J., Beem A. L., Mulder E. J., Derks E. M., Riese H., Willemsen G. A., Bartels M., van den Berg M., Kupper N. H., Polderman T. J., Posthuma D., Rietveld M. J., Stubbe J. H., Knol L. I., Stroet T., van Baal G.C. (2002). Netherlands Twin Register: a focus on longitudinal research. Twin Res. 5:401–406
Boomsma, D. I., Willemsen, G., Dolan, C. V., Hawkley, L. C., and Cacioppo, J. T. (2005). Genetic and environmental contributions to loneliness in adults: The Netherlands twin register study. Behav. Genet. 35(6): 745–752
Brzustowicz L. M., Hodgkinson K. A., Chow E. W., Honer W. G., Bassett A. S. (2000). Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21–q22. Science 288:678–682
Cacioppo J. T., Hawkley L. C., Berntson G. G., Ernst J. M., Gibbs A. C., Stickgold R., Hobson J. A. (2002-a). Lonely days invade the nights: Social modulation of sleep efficiency. Psychol. Sci. 13:385–388
Cacioppo J. T., Hawkley L. C., Crawford L. E., Ernst J. M., Burleson M. H., Kowalewski R. B., Malarkey W. B., Van Cauter E., Berntson G. G. (2002-b). Loneliness and health: Potential mechanisms. Psychosomatic Med. 64:407–417
Cacioppo, J. T., Hughes, M. E., Waite, L. J., Hawkley, L. C., and Thisted, R. A. (in press). Loneliness as a specific risk factor for depressive symptoms: Cross sectional and longitudinal analyses.Psychol. Aging
Camp N. J., Lowry M. R., Richards R. L., Plenk A. M., Carter C., Hensel C. H., Abkevich V., Skolnick M. H., Shattuck D., Rowe K. G., Hughes D. C., Cannon-Albright L. A. (2005). Genome-wide linkage analyses of extended Utah pedigrees identifies loci that influence recurrent, early-onset major depression and anxiety disorders. Am. J. Med. Genet. B (Neuropsychiatr. Genet.) 135B:85–93
Churchill G. A., Doerge R. W. (1994). Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Craddock N., O’Donovan M. C., Owen M. J. (2005). The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J Med Genet 42:193–204
Curtis D., Kalsi G., Brynjolfsson J., McInnis M., O’Neill J., Smyth C., Moloney E., Murphy P., McQuillin A., Petursson H., Gurling H. (2003). Genome scan of pedigrees multiply affected with bipolar disorder provides further support for the presence of a susceptibility locus on chromosome 12q23–q24, and suggests the presence of additional loci on 1p and 1q. Psychiatr. Genet. 13:77–84
Degn B., Lundorf M. D., Wang A., Vang M., Mors O., Kruse T. A., Ewald H. (2001). Further evidence for a bipolar risk gene on chromosome 12q24 suggested by investigation of haplotype sharing and allelic association in patients from the Faroe Islands. Mol. Psychiatry. 6:450–455
Detera-Wadleigh S. D. (1999). Chromosomes 12 and 16 workshop. Am. J. Med. Genet. 88:255–259
Duck S., Pond K., Leatham G. (1994). Loneliness and the evaluation of relational events. J. Soc. Pers. Relat. 11:253–276
Eisemann M. (1984). Contact difficulties and experience of loneliness in depressed patients and non-psychiatric controls. Acta Psychiatr. Scand. 70:160–165
Eisemann M. (1985). Depressed patients and non-psychiatric controls: Discriminant analysis of social environmental variables. Acta Psychiatr. Scand. 71:495–498
Ernst J. M., Cacioppo J. T. (1999). Lonely hearts: Psychological perspectives on loneliness. Appl. Prev. Psychol. 8:1–22
Ekholm J. M., Kieseppa T., Hiekkalinna T., Partonen T., Paunio T., Perola M., Ekelund J., Lonnqvist J., Pekkarinen-Ijas P., Peltonen L. (2003). Evidence of susceptibility loci on 4q32 and 16p12 for bipolar disorder. Hum. Mol. Genet. 12:1907–1915
Ewald H., Flint T., Kruse T.A., Mors O. (2002). A genome-wide scan shows significant linkage between bipolar disorder and chromosome 12q24.3 and suggestive linkage to chromosomes 1p22–21, 4p16, 6q14–22, 10q26 and 16p13.3. Mol. Psychiatr. 7:734–744
Falconer D. S. (1989). Introduction to Quantitative Genetics. London, Longman
Fullerton J., Cubin M., Tiwari H., Wang C., Bomhra A., Davidson S., Miller S., Fairburn C., Goodwin G., Neale M. C., Fiddy S., Mott R., Allison D. B., Flint J. (2003). Linkage analysis of extremely discordant and concordant sibling pairs identifies quantitative-trait loci that influence variation in the human personality trait neuroticism. Am. J. Hum. Genet. 72:879–890
Glaser, B., Kirov, G., Bray, N. J., Green, E., O’Donovan, M. C., Craddock, N. and Owen, M. J. (2005a). Identification of a potential bipolar risk haplotype in the gene encoding the winged-helix transcription factor RFX4. Mol. Psychiatr. 10(10) : 920–927
Glaser B., Kirov G., Green E., Craddock N., Owen M. J. (2005b). Linkage disequilibrium mapping of bipolar affective disorder at 12q23-q24 provides evidence for association at CUX2 and FLJ32356. Am. J. Med. Genet. B (Neuropsychiatr. Genet.) 132:38–45
Green E. K., Elvidge G. P., Owen M. J., Craddock N. (2003). Mutational analysis of two positional candidate susceptibility genes for bipolar disorder on chromosome 12q23–q24: phenylalanine hydroxylase and human LIM-homeobox LHX5. Psychiatr. Genet. 13:97–101
Green E., Elvidge G., Jacobsen N., Glaser B., Jones I., O’Donovan M. C., Kirov G., Owen M. J., Craddock N. (2005). Localization of bipolar susceptibility locus by molecular genetic analysis of the chromosome 12q23-q24 region in two pedigrees with bipolar disorder and Darier’s disease. Am. J. Psychiatr. 162:35–42
Goldsmith S. K., Pellmar T. C., Kleinman A. M., Bunney W. E. (2002). Reducing Suicide: A National Imperative. Washington, DC, National Academy Press
Hamet P., Tremblay J. (2005). Genetics and genomics of depression. Metab. Clin. Exp. 54:10–15
Hawkley, L. C., Browne, M. W., and Cacioppo, J. T. (2005). How can I connect with thee? Psychol. Sci. 16(10) : 798–804
Heijmans, B. T., Beekman, M., Putter, H., Lakenberg, N., van der Wijk, H. J., Whitfield, J. B., Posthuma, D., Pedersen, N. L., Martin, N. G., Boomsma, D. I., and Slagboom, P. E. (2005). Meta-analysis of four new genome scans for lipid parameters and analysis of positional candidates in positive linkage regions. Eur. J. Hum. Genet. 13(10) : 1143–1153.
Heikkinen R., Kauppinen M. (2004). Depressive symptoms in late life: A 10-year follow-up. Arch. Gerontol. Geriatr. 38:239–250
Jones I., Jacobsen N., Green E. K., Elvidge G. P., Owen M. J., Craddock N. (2002). Evidence for familial cosegregation of major affective disorder and genetic markers flanking the gene for Darier’s disease. Mol. Psychiatr. 7:424–427
Kong A., Gudbjartsson D. F., Sainz J., Jonsdottir G. M., Gudjonsson S. A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G., Shlien A., Palsson S. T., Frigge M. L., Thorgeirsson T. E., Gulcher J. R., Stefansson K. (2002). A high-resolution recombination map of the human genome. Nat. Genet. 31:241–247
Lander E., Kruglyak L. (1995). Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat. Genet. 11:241–247
Lewis C. M., Levinson D. F., Wise L. H., DeLisi L. E., Straub R. E., Hovatta I., Williams N. M., Schwab S. G., Pulver A. E., Faraone S. V., Brzustowicz L. M., Kaufmann C. A., Garver D. L., Gurling H. M., Lindholm E., Coon H., Moises H. W., Byerley W., Shaw S. H., Mesen A., Sherrington R., O’Neill F.A., Walsh D., Kendler K. S., Ekelund J., Paunio T., Lonnqvist J., Peltonen L., O’Donovan M. C., Owen M. J., Wildenauer D. B., Maier W., Nestadt G., Blouin J. L., Antonarakis S. E., Mowry B. J., Silverman J. M., Crowe R. R., Cloninger C. R., Tsuang M. T., Malaspina D., Harkavy-Friedman J. M., Svrakic D. M., Bassett A. S., Holcomb J., Kalsi G., McQuillin A., Brynjolfson J., Sigmundsson T., Petursson H., Jazin E., Zoega T., Helgason T. (2003). Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet 73:34–48
Lynch M., Walsh B. (1998). Genetics and Analysis of Quantitative Traits. Sunderland, MA, Sinauer Associates
Maziade M., Roy M. A., Chagnon Y. C., Cliche D., Fournier J. P., Montgrain N., Dion C., Lavallee J. C., Garneau Y., Gingras N., Nicole L., Pires A., Ponton A. M., Potvin A., Wallot H., Merette C. (2005). Shared and specific susceptibility loci for schizophrenia and bipolar disorder: a dense genome scan in Eastern Quebec families. Mol. Psychiatr. 10:486–499
McGuire S., Clifford J. (2000). Genetic and environmental contributions to loneliness in children. Psychol. Sci. 11:487–491
Monks S. A., Kaplan N. L. (2000). Removing the sampling restrictions from family-based tests of association for a quantitative-trait locus. Am. J. Hum. Genet. 66:576–592
Morissette J., Villeneuve A., Bordeleau L., Rochette D., Laberge C., Gagne B., Laprise C., Bouchard G., Plante M., Gobeil L., Shink E., Weissenbach J., Barden N. (1999). Genome-wide search for linkage of bipolar affective disorders in a very large pedigree derived from a homogeneous population in quebec points to a locus of major effect on chromosome 12q23-q24. Am. J. Med. Genet. 88:567–587
Nash M. W., Huezo-Diaz P., Williamson R. J., Sterne A., Purcell S., Hoda F., Cherny S. S., Abecasis G. R., Prince M., Gray J. A., Ball D., Asherson P., Mann A., Goldberg D., McGuffin P., Farmer A., Plomin R., Craig I. W., Sham P. C. (2004). Genome-wide linkage analysis of a composite index of neuroticism and mood-related scales in extreme selected sibships. Hum. Mol. Genet. 13:2173–2182
Neale, M., Boker, S. M., and Maes, H. H. (2003). Mx: Statistical Modelling. 6th edn, VCU Box 900126, Richmond VA 23298: Department of Psychiatry
Nyholt, D. R., Morley, K. I., Ferreira, M. A., Medland, S. E., Boomsma, D. I., Heath, A. C., Merikangas, K. R., Montgomery, G. W., Martin, N. G. (2005). Genomewide significant linkage to migrainous headache on chromosome 5q21. Am. J. Hum. Genet. 77(3):500–512
Peplau L. A., Perlman D. (1982). Perspective on loneliness. In: Peplau L. A., Perlman D. (eds), Loneliness: A Sourcebook of Current Theory, Research and Therapy. New York, Wiley, pp. 1–20
Rook K.S. (1988). Towards a more differentiated view of loneliness. In: Duck S. (eds), Handbook of Personal Relationships: Theory, Research and Interventions. Chichester, England, Wiley, pp. 571–590
Russell D., Peplau L. A., Cutrona C. E. (1980). The revised UCLA loneliness scale: Concurrent and discriminant validity evidence. J. Personality Soc. Psychol. 39:472–480
Segrin C. (1998). Interpersonal communication problems associated with depression and loneliness. In: Andersen P. A., Guerrero L. K. (eds), Handbook of Communication and Emotion: Research, Theory, Applications, and Contexts. San Diego, CA, Academic Press, Inc, pp. 215–242
Segurado R., Detera-Wadleigh S. D., Levinson D. F., Lewis C. M., Gill M., Nurnberger J. I. Jr, Craddock N., DePaulo J. R., Baron M., Gershon E. S., Ekholm J., Cichon S., Turecki G., Claes S., Kelsoe J. R., Schofield P. R., Badenhop R. F., Morissette J., Coon H., Blackwood D., McInnes L. A., Foroud T., Edenberg H. J., Reich T., Rice J. P., Goate A., McInnis M. G., McMahon F. J., Badner J. A., Goldin L. R., Bennett P., Willour V. L., Zandi P. P., Liu J., Gilliam C., Juo S. H., Berrettini W. H., Yoshikawa T., Peltonen L., Lonnqvist J., Nothen M. M., Schumacher J., Windemuth C., Rietschel M., Propping P., Maier W., Alda M., Grof P., Rouleau G. A., Del-Favero J., Van Broeckhoven C., Mendlewicz J., Adolfsson R., Spence M. A., Luebbert H., Adams L. J., Donald J. A., Mitchell P. B., Barden N., Shink E., Byerley W., Muir W., Visscher P. M., Macgregor S., Gurling H., Kalsi G., McQuillin A., Escamilla M. A., Reus V. I., Leon P., Freimer N. B., Ewald H., Kruse T. A., Mors O., Radhakrishna U., Blouin J. L., Antonarakis S. E., Akarsu N. (2003). Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: Bipolar disorder. Am. J. Hum. Genet. 73:49–62
Sham P. (1998). Statistics in Human Genetic. Arnold Publishers, London
Shaver P.R., Brennan K.A. (1991). Measures of depression and loneliness. In: Robinson J.P., Shaver P.R., Wrightsman L.S. (eds), Measures of Personality and Social Psychological Attitudes. Vol. 1 San Diego, Academic Press, pp. 195–290
Shink E., Harvey M., Tremblay M., Gagne B., Belleau P., Raymond C., Labbe M., Dube M. P., Lafreniere R. G., Barden N. (2005a). Analysis of microsatellite markers and single nucleotide polymorphisms in candidate genes for susceptibility to bipolar affective disorder in the chromosome 12Q24.31 region. Am. J. Med. Genet. B (Neuropsychiatr. Genet.) 135B:50–58
Shink E., Morissette J., Sherrington R., Barden N. (2005b). A genome-wide scan points to a susceptibility locus for bipolar disorder on chromosome 12. Mol. Psychiatr. 10:545–552
Stenger J. E., Xu H., Haynes C., Hauser E. R., Pericak-Vance M., Goldschmidt-Clermont P. J., Vance J. M. (2005) Statistical Viewer: a tool to upload and integrate linkage and association data as plots displayed within the Ensembl genome browser. BMC Bioinformatics 6:95
Toyota T., Watanabe A., Shibuya H., Nankai M., Hattori E., Yamada K., Kurumaji A., Karkera J. D., Detera-Wadleigh S. D., Yoshikawa T. (2000). Association study on the DUSP6 gene, an affective disorder candidate gene on 12q23, performed by using fluorescence resonance energy transfer-based melting curve analysis on the LightCycler. Mol. Psychiatr. 5:489–494
Weiss R. S. (1973). Loneliness: The Experience of Emotional and Social Isolation. Cambridge, MIT Press
Wenz F. V. (1977). Seasonal suicide attempts and forms of loneliness. Psychol. Rep. 40:807–810
Wilcox M. A., Faraone S. V., Su J., Van Eerdewegh P., Tsuang M. T. (2002). Genome scan of three quantitative traits in schizophrenia pedigrees. Biol. Psychiatr. 52:847–854
Yi N., Xu S., George V., Allison D. B. (2004). Mapping Multiple Quantitative Trait Loci for Ordinal Traits. Behav. Genet. 34:3–15
Yuan B., Vaske D., Weber J. L., Beck J., Sheffield V. C. (1997) Improved set of short-tandem-repeat polymorphisms for screening the human genome. Am. J. Hum. Genet. 60:459–460
Zubenko G. S., Maher B., Hughes H. B. 3rd., Zubenko W. N., Stiffler J. S., Kaplan B. B., Marazita M. L. (2003). Genome-wide linkage survey for genetic loci that influence the development of depressive disorders in families with recurrent, early-onset, major depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 123B: 1–18
Acknowledgments
Supported by NWO (Netherlands Organization for Scientific Research) grants 575-25-006, 904-61-090, 985-10-002, 904-61-193 and NWO/SPI 56-464-14192; National Supercomputing Facilities (NCF) and the National Institute of Aging Grant PO1 AG18911 (Social isolation, loneliness, health, and the aging process). Dr. Posthuma is supported in part by GenomEUtwin grant (EU/QLRT-2001-01254). Genotyping was carried out by Profs. Weber (Centre for Medical Genetics in Marshfield) and Slagboom (LUMC). We would like to thank E. Suchiman and N. Lakenberg for DNA isolation and genotyping and Drs. A.L. Beem and J.J. Hottenga for statistical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Boomsma, D.I., Cacioppo, J.T., Slagboom, P.E. et al. Genetic Linkage and Association Analysis for Loneliness in Dutch Twin and Sibling Pairs Points to a Region on Chromosome 12q23–24. Behav Genet 36, 137–146 (2006). https://doi.org/10.1007/s10519-005-9005-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-005-9005-z